|
|
Clinical Pharmacology of SSRI's
2 - Rational Drug Discovery and SSRIs |
|
|
|
|
|
Over the past decade, tremendous strides have been made in
the treatment of major depression due to the ability to rationally
develop psychiatric medications.215
In the last 10 years, 6 new medications have been marketed
as antidepressants in
the U.S. (Table 2.1):
| TABLE 2.1 Major Classes of
Antidepressants Defined by Principal Mechanisms of Action |
- Combined NE and 5-HT uptake inhibition, plus effects
on multiple other neuroreceptors and fast sodium channels
Tertiary amine tricyclic antidepressants (TCAs)
- 5-HT uptake inhibition Serotonin selective reuptake
inhibitors (SSRIs)
- NE uptake inhibition Secondary amine TCAs
- Combined NE and 5-HT uptake inhibition Venlafaxine
- 5-HT2 receptor blockers and 5-HT uptake inhibition
Nefazodone (phenylpiperazine)
- DA and NE uptake inhibitors Bupropion (aminoketones)
- Monoamine oxidase inhibitors (MAOIs) Nonselective
and irreversible Selective and/or reversible (RIMAs)
|
- Bupropion
- Fluoxetine
- Sertraline
- Paroxetine
- Venlafaxine
- Nefazodone
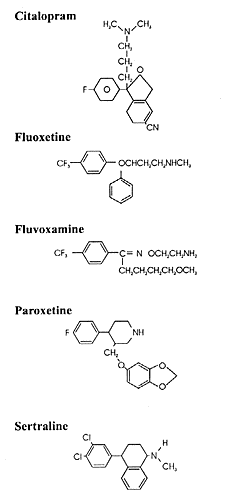
Three of these drugs (fluoxetine, paroxetine and sertraline),
together with two other drugs (citalopram and fluvoxamine)
marketed as antidepressants elsewhere in the world, form the
class known as selective serotonin reuptake inhibitors (SSRIs)
(Figure 2.1). For many physicians, this
class has supplanted tricyclic antidepressants (TCAs) as the
antidepressant of first choice due to their greater safety
and tolerabilty, coupled with comparable efficacy.
The rapid expansion in the number of antidepressants and
the change in what is considered first-line therapy has been
accompanied by a substantial amount of commercial claims and
counterclaims. This situation can be confusing, even for physicians
who specialize in clinical psychopharmacology, and even more
so for the general physician who must also contend with developments
in other therapeutic areas. Therefore, this book will explain
the differences between the SSRIs and the TCAs, which were
the mainstay of pharmacotherapy for major depression for many
years, and discuss the clinically
important similarities and differences between members of
the SSRI class.
The development of SSRIs occurred over a relatively short
interval of time. The first SSRI marketed was zimelidine by
Astra. Unfortunately, several cases of Guillain-Barre syndrome
were associated with the use of this drug and led to its withdrawal
from the market. Nonetheless, five SSRIs were eventually launched
successfully in multiple countries around the world. Each
was developed by a different company:
- Citalopram by Lundbeck
- Fluvoxamine by Solvay
- Fluoxetine by Lilly
- Paroxetine by SmithKline-Beecham
- Sertraline by Pfizer
|
Parenthetically, while fluvoxamine is marketed
as an antidepressant in many parts of the world, it is marketed
only for obsessive-compulsive disorder in the U.S. Citalopram,
while marketed in several countries in the world as an antidepressant,
is not yet available in the U.S. Fluoxetine was the first
SSRI marketed in the United States in 1988. Table
10.2 (in the Appendix) lists the SSRIs that are available
in various countries.
The fact that the five SSRIs were produced by five different
companies is a testimony to the shift from a discovery process
dependent on chance observation to a process of rational drug
development. Understanding rational drug development is pivotal
to understanding the clinical pharmacology of SSRIs.
How the SSRIs were developed is a scientific success story.
The SSRIs are the first rationally designed class of psychotropic
medications and, hence, have launched a new era in psychotropic
drug development. The strategy behind rational drug development
is to design a new drug that is capable of affecting a specific
neural site of action (SOA) (eg, uptake pumps, receptors)
while avoiding effects on other SOAs. The goal in such development
is to produce agents that are more efficacious, safer and
better tolerated than older medications (Table 2.2).215
This enhanced safety profile includes a reduced likelihood
of pharmacodynamically mediated adverse drug-drug interactions
by avoiding affects on SOAs that are not essential to the
intended outcome (eg, antidepressant efficacy).
A few general comments about what a drug must do to produce
a specific clinical effect may be helpful to put this book
in perspective. A drug must act on an SOA that is physiologically
relevant to the effect (Figure 2.2). That SOA may be, by way
of example but not limited to, an uptake pump, an enzyme,
or a receptor. The drug "recognizes" and binds to
that SOA. The activation or inhibition of a specific site
is termed the drug's mechanism of action (MOA). For example,
a drug may be an agonist or antagonist at a specific serotonin
receptor.
A given drug may affect one or more SOAs over its clinically
relevant dosing range and, therefore, may produce
multiple and different clinical effects, some desired and
some not. Drugs that affect multiple SOAs are more characteristic
of drugs developed based on chance discovery, whereas the
goal of rational drug development is to produce drugs with
a more limited range of effects (Table 2.2).
Prior to the SSRIs, all psychotropic medications were the
result of chance observation (Table 2.3). Lithium came from
studies looking for putative endogenous psychomimetic substances
excreted in the urine of psychotic patients.51
The phenothiazines came from a search for better preanesthetic
agents.154 The TCAs
were the result of an unsuccessful attempt to improve on the
antipsychotic effectiveness of phenothiazines.148
The monoamine oxidase inhibitors (MAOIs) came from a failed
attempt to develop effective antitubercular medications.63
The first studies of benzodiazepines
were unsuccessful attempts to treat patients with schizophrenia.
| TABLE 2.3 The Evolution of
Psychopharmacology |
- First generation discovered by chance (eg, tricyclic
antidepressants)
- Advanced generations discovered by design based
on molecular targeting (eg, serotonin selective reuptake
inhibitors)
|
Despite these initial failed attempts to use these various
drugs therapeutically, astute clinical investigators recognized
their therapeutic value in other conditions: lithium for manic-depression,
phenothiazines for psychotic disorders, TCAs and MAOIs for
major depression, and benzodiazepines for anxiety disorders.
This chance discovery process is not unique to psychiatry,
but instead has been the universal first step in any therapeutic
area.210 Prior to
such first steps, too little knowledge of the biology underlying
illnesses existed to permit a more rational
approach to drug development. However, these first drugs played
an important role in providing the first insights into the
pathophysiology underlying the illness or, at least, underlying
drug responsiveness.
Molecular targeting is the essence of rational drug development.211
In this approach, the specific target(s) of interest is a
fundamental brain mechanism believed to be important in the
pathophysiology underlying a specific psychopathologic condition
or psychiatric syndrome (eg, major depression). This SOA may
be the neuronal uptake pump for a neurotransmitter, a specific
neurotransmitter receptor subtype, or a subunit of an ion
channel (Figure 2.2).
As was the case with each of the SSRIs, the new molecular
entity is developed to stereospecifically interact with the
target of interest. At the same time, the molecule is structurally
modified so that it does not interact with other targets that
mediate unwanted effects (eg, peripheral anticholinergic effects).
Through this systematic approach, a new candidate drug is
selected for clinical testing to support registration for
marketing. This type of rational drug development is now possible
in psychiatry because of the improved understanding of central
and peripheral mechanisms of action (MOAs) relevant to both
desired and undesired central and peripheral
effects.
| FIGURE 2.3 Standard and New
Generation Antidepressants Mechanisms of Action |
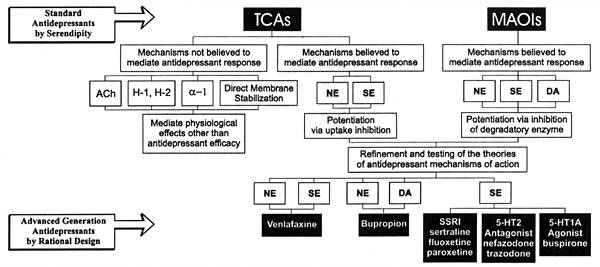 |
Antidepressant pharmacotherapy is the first area in psychopharmacology
to have benefitted significantly from such targeted development.215
Figure 2.3 illustrates the evolution of antidepressants over
the past three decades. TCAs and MAOIs were the first successful
antidepressants, but their antidepressant properties were
discovered by chance, as discussed above. Nonetheless, this
chance discovery was important. First, these drugs provided
the first scientifically proven treatments for major depression
and demonstrated that major depression was amenable to medical
intervention just as other medical conditions, such as hypertension
and diabetes. Second, they served as roadmaps to improve our
understanding of the MOAs, mediating both their desired antidepressant
effects and undesired effects. This information was critical
to the rational drug development efforts that followed and
led to the SSRIs and other new antidepressants.
In the case of major depression, TCAs and MAOIs implicated
the potentiation of neurotransmission in one or more than
one central biogenic amine neural system as potential MOAs
responsible for their antidepressant efficacy. That finding,
coupled with improved means of isolating and studying the
effects of drugs on specific neural mechanisms, led to the
development of the SSRIs.
The nature of older chance-discovery drugs is that they have
many clinical effects either because they affect an SOA with
broad implications for organ function (eg, MAOIs that affect
an enzyme responsible for the degradation of four major neurotransmitters)
or because they affect multiple SOAs (eg, TCAs). Such drugs
typically have:
- Narrow therapeutic indices
- Poor tolerability profiles
- Potential for causing multiple types of pharmacodynamic
interactions with a wide variety of concomitantly prescribed
medications
The SSRIs were developed based on the knowledge gained from
studying the effects of the TCAs and the techniques developed
in basic neuroscience research to isolate and study the effects
of drugs on specific neural SOAs (eg, uptake pumps, receptors).
In the case of the SSRIs, each was the product of a similar
development strategy in which the goal was to produce a drug
capable of inhibiting the neuronal uptake pump for serotonin,
a property shared with the TCAs, but without affecting the
various other neuroreceptors (ie, histamine,
acetylcholine, and a-adrenergic receptors) or fast sodium
channels, affected by the TCAs. Actions on these latter sites
are responsible for many of the safety and tolerability problems
of the TCAs.220,221
The fact that SSRIs were designed to avoid affecting these
other SOAs explains many of the pharmacological differences
between the SSRIs and the TCAs (see
Section 4) and explains the similarities among the SSRIs
(see Section 5). In many ways,
the SSRIs are to psychiatry as b-blockers are to internal
medicine.
In contrast to rational development, chance discovery is
usually dependent on the drug's having a large signal-to-noise
ratio (ie, a big clinical effect or multiple clinical effects).
Unfortunately, this fact means that chance-discovery drugs
typically will produce a number of undesired, as well as desired,
effects and will have a narrower therapeutic index in comparison
with a drug that was rationally developed to affect only the
SOA(s) necessary to produce the desired response.
This issue can be readily understood by examining the pharmacology
of TCAs that has served as the cornerstone of antidepressant
pharmacotherapy for almost 30 years. TCAs affect multiple
SOAs over a relatively narrow concentration range so that
patients are likely to experience multiple effects while taking
these medications.34,66,221,225
Some MOAs of TCAs (ie, the inhibition of the fast sodium channels)
can cause potentially serious effects on cardiac conduction
and occur at concentrations only an order of magnitude higher
than the concentration needed to inhibit the neuronal uptake
pumps for norepinephrine and serotonin, the putative MOAs
mediating the antidepressant effects of TCAs. This fact explains
why an overdose of TCAs of only 5 to 10 times their therapeutic
dose can cause serious toxicity and why patients who have
a slow clearance rate for these drugs can develop serious
adverse effects on routine doses due to the accumulation of
toxic concentrations.228
To put this issue with TCAs in perspective, Table 2.4 illustrates
the cocktail of drugs, each having only one predominant MOA,
that would have to be given to a patient to reproduce the
effects that occur in a patient receiving a tertiary amine
TCA, such as amitriptyline. Obviously, the problem with amitriptyline
is that the patient has to experience a large number of effects
to receive the benefit of the mechanism
that mediates antidepressant response.
| TABLE 2.4 TCA (Amitriptyline)
Polypharmacy in a Single Pill |
| Drug |
Action |
| Chlorpheniramine |
Histamine-1 receptor blockade |
| Cimetidine |
Histamine-2 receptor blockade |
| Benztropine |
Acetylcholine receptor blockade |
| Desipramine |
Norepinephrine uptake inhibition |
| Sertraline |
Serotonin uptake inhibition |
| Nefazodone |
5-HT2 receptor blockade |
| Prazosin |
NE-a1
receptor blockade |
| Yohimbine |
NE-a2
receptor blockade |
| Quinidine |
Direct membrane stabilization |
| The multiple actions
of amitriptyline are listed in descending order of potency
(ie, histamine -1 receptor blockade is the most potent,
whereas direct membrane stabilization is the least.) |
The issue of multiple MOAs over a narrow concentration range
is further complicated by the fact that there is a large interindividual
variability in the clearance rates of TCAs, even in physically
healthy individuals.213
The variability is even larger when dealing with the elderly,
the medically ill, and patients on concomitant
medications that can either induce or inhibit the clearance
of these drugs. With TCAs, patients can have numerous types
of adverse effects ranging from nuisance problems (eg, dry
mouth) to serious toxicity (eg, seizures, cardiac arrhythmias).
Patients who clear the drugs slowly may experience the latter
due to the accumulation of excessive concentrations despite
being on conventional doses.
This situation is made even more complicated because the
early signs of TCA-induced toxicity can mimic worsening of
major depression so that the physician may unfortunately respond
by increasing rather than reducing the dose.227
These facts considered together have made therapeutic drug
monitoring (TDM), at least once during early treatment (at
the end of the first week of treatment with a stable dose),
a standard aspect when prescribing TCAs.221,228
Using the TDM results, rational dose adjustment can then be
made to compensate for the intraindividual differences in
clearance rate, and thus ensure that the patient will be treated
with a dose that will achieve a concentration that is optimal
for most patients with regard
to efficacy, safety and cost effectiveness.
| FIGURE 2.4 In Vitro
Potency of Amitriptyline as a Representative Tricyclic
Antidepressant for Different Sites of Action and Related
Mechanisms of Action |
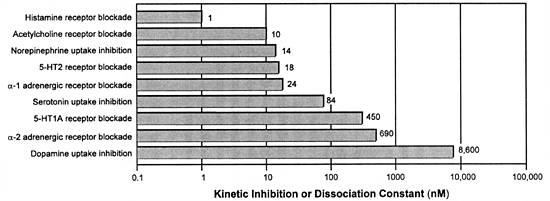 |
| Adapted from references: 34,
66 |
Unfortunately, TDM-driven dose adjustment does not substantially
improve the tolerability of TCAs because MOAs for producing
adverse effects (eg, those mediated by histamine or muscarinic
receptor blockade) are more potent and hence occur at lower
concentrations than their presumed MOAs underlying their antidepressant
efficacy (ie, inhibiting the neuronal uptake for norepinephrine
and serotonin) (Figure 2.4). Hence, patients who are sensitive
to a given MOA may experience discomforting adverse effects
even at concentrations that are subtherapeutic for treating
major depression. That problem has been addressed by rational
drug development of new drugs (eg, SSRIs) with a much wider
gap between their potency for an effect on the desired versus
undesired targets. As a result, we now have
seven major classes of antidepressants based on putative MOAs
mediating antidepressant response (Table
2.1).
The SSRIs were all developed to have a similar MOA: the potentiation
of serotonin (5-HT) by the inhibition of its neuronal uptake
pump. As such, all SSRIs have common 5-HT agonistic effects
that appear to mediate both their desired (eg, antidepressant
efficacy) and undesired (eg, sexual dysfunction) reactions.
As a class, SSRIs are considerably more selective in comparison
to TCAs in terms of their central nervous system MOAs, but
differ in other clinically important ways, as will be discussed
in detail in this book (see Sections
6 through 8).
The reason to choose serotonin uptake inhibition as the desired
MOA is based on the emerging understanding of the role of
serotonin in the brain as well as on the pharmacology of TCAs
and MAOIs. From a phylogenetic standpoint, serotonin is one
of the oldest neurotransmitters.255
It is found in such relatively simple organisms as jellyfish.
In the human brain, serotonin-containing neurons are highly
localized in specific clusters in the brainstem and spinal
cord.271 From these
sites, the cells send out axons that end in serotonin-containing
terminals innervating the diverse areas throughout the brain.
These regions include:
- Spinothalamic pain fibers
- Brainstem
- Cerebellum
- Hypothalamus
- Basal ganglia
- Neocortex
This anatomy explains why serotonin is implicated in so many
brain functions including:
- Pain perception
- Sleep
- Thermal regulation
- Appetite
- Gut regulation
- Balance
- Reproductive function
- Motor function
- Higher cognitive function
- Sensory interpretation
Given these diverse responsibilities, dysfunction of serotonin
neurons have been implicated in a wide variety of diseases,
including major depression. For the same reason, serotonin-active
drugs can have many different clinical effects by virtue of
their physiological effects on diverse brain regions. This
anatomy explains why even "selective" drugs such
as SSRIs can produce so many diverse clinical effects (eg,
nausea, a feeling of incoordination, suppression of REM sleep,
decreased libido, akathisia) as well as being useful in such
apparently disparate disorders as major depression, anxiety
disorders, pain disorders, and premature ejaculation. While
SSRIs are "selective" in terms of affecting the
neuronal uptake pump for serotonin, this action affects a
multitude of specific postsynaptic serotonin receptors (eg,
5-HT1A, 5-HT1D, 5-HT2A, 5-HT2C, and 5-HT3) which, in turn,
affects a multitude of neural systems.128
Chirality
Although all SSRIs are products of rational drug development,
one of the major goals of such a development was not realized
with two of the SSRIs due to the phenomenon of chirality:
that goal was to produce a drug that is a single molecule
with a precise, limited (or focused) range of pharmacological
actions. If the molecule has an asymmetrical carbon, then
it exists in enantiomeric forms (ie, chirality). As can be
seen in Figure 2.1, all of the SSRIs
except fluvoxamine have an asymmetrical
carbon. However, only one enantiomer of paroxetine and sertraline,
respectively, is contained in the marketed formulation of
these two drugs. In contrast, citalopram and fluoxetine are
marketed as the racemates of their two enantiomers. Hence,
patients on these two SSRIs achieve plasma and tissue levels
of each enantiomer and their respective metabolites, which
are also enantiomers.
This fact raises the question of whether there are substantial
differences in the pharmacodynamics and pharmacokinetics of
these enantiomers and whether such differences contribute
in a meaningful way to the variance in drug response among
different patients.12
If the different enantiomers have meaningful differences in
their therapeutic ratios, one enantiomer can contribute disproportionately
to adverse consequences relative to therapeutic benefit. The
presence of enantiomers complicates the use of TDM in both
research and clinical practice since many assays will not
distinguish between the two enantiomeric forms of a drug.
If there are meaningful differences in their pharmacodynamics
and pharmacokinetics, that fact can add substantial "noise"
to such results and thus confound their interpretation.
As is the case with the enantiomers of citalopram and fluoxetine,
there is often limited data on their relative pharmacodynamics
and pharmacokinetics to answer these questions. A summary
of that data for these two SSRIs follows.
The racemic mixture of citalopram produces racemic desmethylcitalopram
and didesmethylcitalopram. The S-enantiomers are potent
and selective inhibitors of serotonin uptake in contrast to
the relatively inactive corresponding R-enantiomers.130
The active S-enantiomer of citalopram is generally
only one-third of the total citalopram plasma level under
steady-state conditions.242
However, there is variability in this ratio among different
patients that may be characteristic of patients
genetically deficient in cytochrome P450 (CYP) 2C19.242
CYP 2C19 enzyme is the principal enzyme responsible for the
metabolism of citalopram.254
This variability in the ratio of the active to the relatively
inactive enantiomer can contribute to variability in response
to the drug among different patients. Given the relative levels
and activity of the enantiomers of citalopram and its metabolites,
studies attempting to correlate plasma levels of citalopram
with serotonin mediated effects should report on the levels
of each enantiomer or should focus on the levels of S-citalopram.
Racemic fluoxetine produces racemic norfluoxetine. While
S-fluoxetine, R-fluoxetine, and S-norfluoxetine
are potent and selective inhibitors of serotonin uptake in
vitro and in vivo, that is not true for R-norfluoxetine.98,290,291
Under steady-state conditions, the plasma levels of racemic
fluoxetine and norfluoxetine are comparable.189
Thus, studies attempting to correlate the plasma levels of
fluoxetine and norfluoxetine should ideally take into account
the relative inactivity of the R-norfluoxetine in terms
of the inhibition of serotonin uptake.
The R-enantiomers of fluoxetine and norfluoxetine
are also weaker inhibitors of CYP 2D6 than are the S-enantiomers.267
Thus, failure to distinguish between these enantiomers in
studies attempting to correlate plasma levels of fluoxetine
and norfluoxetine with the inhibition of the metabolism of
CYP 2D6-dependent substrates will hamper the ability to establish
such a relationship.
Tables summarizing the above data are found in the sections
dealing with the effects of the SSRIs on neural mechanisms
(Section 3, Table 3.4) and
on CYP enzymes (Section 8,
Table 8.11), respectively. There may be other important
differences in the pharmacodynamics and pharmacokinetics of
the enantiomers of these two SSRIs which
are not known at this time. There is little active research
ongoing in this area; therefore, knowledge of these enantiomers
may not expand appreciably in the near future. This discussion
should be kept in mind as a caveat when reading the rest of
this book. Unless specified otherwise, the data in this book
on in vitro and in vivo studies with citalopram
and fluoxetine were done with the racemic mixtures, and the
plasma and tissue levels reported of the parent compound and
the metabolites are the combined levels of their enantiomeric
forms.
|