|
|
| If Lack of Concentration Didn't Cause the Fall, What Did? |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, November 1996, 364-367
|
In the question and answer period following a lecture, I am frequently asked by a physician in the audience: "Why didn't my patient get better? I had him/her on the usually effective dose and even had therapeutic plasma drug levels." It is a tough but fair question. It emphasizes that physicians are less interested in what happens to the "usual" patient than in what happens to their specific patients. There is no single answer to this question; rather, a series of factors must be systematically explored.
In this column, I will discuss these factors and our increasing ability to understand and make appropriate adjustments for the patient who falls off the dose-response curve despite being on the "usually effective" dose and achieving "usually effective" plasma drug levels. To provide the backdrop for this discussion, I will first briefly review some points covered in more detail in earlier columns.
The goal of medicine is either to prevent illness or restore health when illness strikes. To accomplish these goals, we want safe, effective, and predictable treatments, but predictability is often thwarted to varying degrees by interinidividual variability in response. However, science is improving our understanding of the reasons for interindividual variance in response and is developing ways to predict such variance so that adjustments can be made prospectively rather than after the patient has had a less than optimal outcome.
Such developments are replacing the approach of beginning treatment with the "usual" dose and then adjusting the dose based on clinical assessment of response. That approach, while time honored and still common, is often inefficient and inaccurate, since a sizable percentage of patients are outliers on the usual dose-response curve. Tolerability outliers either 1) do not tolerate doses that are usually well tolerated or 2) tolerate doses that are usually intolerable or even potentially toxic. Efficacy outliers either 1) respond to doses that are usually ineffective or 2) do not respond to usually effective doses (see my January 1996 column).
Factors That Contribute to Variance in Response
The following equation is fundamental to understanding a specific patient's response to a medication and the variance in response among different patients:
Effect = pharmacodynamics
x pharmacokinetics
x underlying biology
Pharmacodynamics refers to what the drug does to the body via its effect on its site(s) of action. Pharmacokinetics refers to what the body does to the drug (i.e., absorption, distribution, metabolism, and elimination). Underlying biology refers to biological differences among patients that can shift the dose-response curve. Such differences can be due to a variety of factors, including genetics, gender, age (i.e., maturational status), disease (i.e., organ impairment), and environment (e.g., concomitantly prescribed medications, diet).
Pharmacodynamic and Pharmacokinetic Factors
Pharmacodynamic and pharmacokinetic factors in the average patient determine the "usual" response in the average patient. This works because our patients belong to the same species and thus are more alike than different. However, humans are not exactly alike, which is why the third factor, underlying biology, is important in understanding the specific rather than the "usual" response. Underlying biology is expressed through the variance between individuals in pharmacodynamics and pharacokinetics.
Nonresponse may mean that the mechanism of action (i.e., pharmacodynamics) of the drug is not relevant to the pathophysiology underlying the patient's illness. The purpose of differential diagnosis is to account for biologically important variance among patients who have the same symptomatic presentation but whose conditions have different pathophysiologies and/or pathoetiologies.
Nonresponse or excessive adverse effects may mean that the site of action is not optimally engaged for pharmacokinetic reasons. If a patient clears the drug considerably more rapidly than most patients, he or she can achieve a drug concentration too low to engage the site of action sufficiently to correct the pathophysiology. Conversely, the patient may clear the drug much more slowly than most patients and develop too high a concentration, which engages the site of action excessively and results in adverse effects that overwhelm the therapeutic benefit. Detection and correction of these causes of interindividual variability in response is the underlying rationale for therapeutic drug monitoring (See my May and July 1996 columns). A major determinant of interindividual variance in drug clearance is genetically determined differences in the functional activity of specific cytochrome P450 (CYP) enzymes (see November 1995 and March 1996 columns).
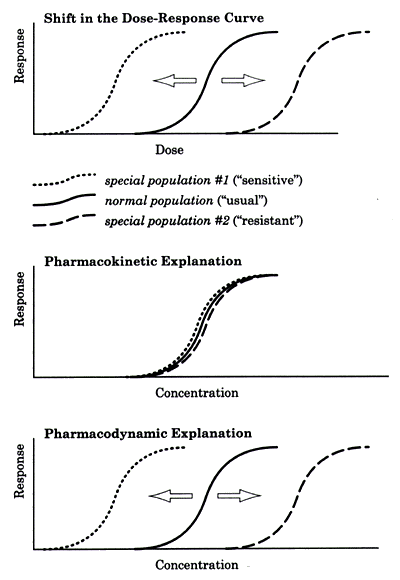 | | Figure 1 - Possible explanations for a shift in the dose-response curve in special populations. |
Three Populations: the Usual, the Sensitive, and the Resistant
The top graph in Figure 1 illustrates the variance that is frequently experienced in clinical practice. This graph shows a population treated with different doses of a medication to establish the dose-response curve. There are three subpopulations. The largest percentage of patients belong to the middle group -- the "normal" population -- whose data represent the "usual" dose-response curve. Another group is "more sensitive" to the drug and their dose-response curve is shifted to the left. The third group is "less sensitive" to the drug, reflected by the shift to the right of their dose-response curve.
These differences might be fully explained by pharmacokinetic differences among the three groups in terms of drug clearance, as illustrated in the middle graph in Figure 1. The group that was shifted to the left was "more sensitive" because they clear the drug more slowly than the usual population and their prescribed dose is thus functionally higher. Conversely, the group that was shifted to the right was "less sensitive" because they clear the drug more rapidly than the usual population and thus their prescribed dose is functionally lower. When the data for these three groups are plotted as a function of concentration, they become superimposable. These three populations are therefore not truly different with regard to their "sensitivity" to the mechanism of action of the drug but simply differ in their ability to clear the drug.
The bottom graph in figure 1 illustrates a situation in which the above explanation does not apply. The three populations do not disappear when response is plotted as a function of drug level rather than dose. Instead the three groups remain separate and distinct because they are in fact different with regard to sensitivity to the mechanism of action of the drug.
This outcome raises several questions. What accounts for the difference in the sensitivity of these populations? How can we detect this difference? And can we detect it in advance so that we can take this difference into account when selecting the medication and dose for the patient?
Response Variance Due to Pharmacodynamics
As can be deduced from equation 1, response to medication at a different plasma drug level indicates a difference in mechanism of action. Until recently, it was erroneously thought that substantially higher doses of selective serotonin reuptake inhibitors were needed to treat patients with obsessive-compulsive disorder (OCD) than patients with major depressive disorder (MDD). Fixed dose studies have not confirmed this difference, but let us assume for the sake of discussion that fixed dose studies actually did substantiate such a difference. The question would be why?
The simplest explanation would be a pharmacokinetic difference between patients with OCD and those with MDD that causes the patients with OCD to clear the drugs much faster than those with MDD and thus need higher doses to achieve the same therapeutic plasma levels (a scenario illustrated in the middle graph in Figure 1). Conceivably, there could be a link between a genetically determined difference in the CYP enzyme that mediates the metabolism of SSRIs and the etiology of OCD, perhaps simply a linkage phenomenon. Alternatively, CYP enzymes are located in the brain as well as in the intestines and liver; hence there might be a causal link between a change in CYP enzyme activity and OCD. One advantage of a pharmacokinetic explanation is that it is easy to test. One simply measures the drug levels achieved in the two different populations to assess whether there is a difference. However, a pharmacokinetic explanation would have been unlikely in this case since different SSRIs are metabolized by different CYP enzymes and there would have had to have been differences in the activity of multiple CYP enzymes between patients with OCD and MDD.
Different Site of Action
Once a pharmacokinetic explanation is ruled out, the search would focus on a difference in pharmacodynamics. One possible explanation is that different drug levels are required because the site of action that mediates the efficacy of the drugs in OCD is different than in MDD. Drugs can affect several sites of action in different conditions. For example, lower doses and hence lower levels of tertiary amine tricyclic antidepressants such as amitriptyline can be used to treat enuresis than are needed to treat MDD. The reason is that amitriptyline is much more potent as a muscarinic acetylcholine blocker than as an inhibitor of norepinephrine and serotonin uptake pumps. The former mechanism is most likely responsible for the anti-enuresis efficacy whereas the latter is presumed to be responsible for antidepressant efficacy. Today enuresis is seen as something quite different than MDD, but only a few years ago it was viewed as "masked" depression, based partly on the fact that it could be treated with an antidepressant such as amitriptyline.
Same Site Yet Different
However, a pharmacodynamic explanation does not require postulating a different site of action. Instead, a mutation of the site of action that affects the drug's binding affinity for the site could be the culprit. Such mutations are analogous to mutations in the CYP enzymes responsible for the genetic polymorphisms that account for differences in elimination rates of drugs in different people. The sites of action of drugs are proteins too and are thus under genetic control.
Although our knowledge of mutations of sites of action lags substantially behind our knowledge of CYP enzyme mutations, some have already been identified along with their role in explaining the differential response to drugs in different subpopulations. For example, higher doses of ß-blockers are required to treat hypertension in blacks than in whites; and even with higher doses, their efficacy is less in blacks than in whites. Genetically determined differences in ß-receptors between blacks and whites have now been identified that may account for these differences. The genetically determined differences in the ß-receptor alter the binding affinity of ß-blockers to the receptor. Higher levels of the drugs are thus required to achieve the same degree of receptor occupancy in patients with one form of the receptor than the other.
In addition to explaining why different levels of the drugs are needed, these mutations may also be important in the etiology of the disease itself. The mutations may not only affect the binding affinity of drugs to the receptor but also how functional the receptor is, which in turn may be important in the pathophysiology and pathoetiology of the disease. Mutations have been identified in serotonin and dopamine receptors and in the serotonin transporter protein (i.e., the serotonin uptake pump) that can alter the concentration of drugs such as SSRIs that are needed to block such sites of action to a comparable degree. Such mutant receptors are being discovered at an increasing rate as a result of advances in molecular biology. Understanding and identifying such mutations is one important goal of the human genome project.
Where Is This Research Leading?
As one can see, this research folds back on itself. Identifying a difference in the dose-response curve between populations or between specific conditions suggests a difference in one of the three factors in equation 1. Part of the underlying biological variance factor in equation 1 can be genetically determined differences in either the site of action, which mediates the pharmacodynamics of the drugs, or in the CYP enzymes, which determine the pharmacokinetics of the drug in terms of its rate of elimination from the body (i.e., its clearance). A mutation in the site of action can alter the concentration of the drug needed to achieve a specified degree of receptor occupancy, whereas a mutation in the CYP enzyme can alter the dose needed to achieve that concentration. These factors can explain differences in dose-response curves among different patients. They will also yield even more fundamental advances in our understanding of the illnesses themselves and in our ability to treat and eventually even prevent them.
In the not too distant future, physicians will be able to genotype their patients for such differences. This information could be stored in a handheld computer and used to select a drug and individualize the dose to treat the patient safely and effectively by taking the guess work out of trying to adjust for interindividual differences in pharmacodynamics and pharmacokinetics.
Suggested Readings
- Asghari V, Schoots O, Van Katz S, et al. Dopamine D4 receptor repeat: Analysis of different native and mutant forms of the human and rat genes. Mol Pharmacol 1994;46:363-73.
- Green PC, et al. A polymorphism of the human beta 2-adrenergic receptor within the fourth transmembrane domain alters ligand binding and functional properties of the receptor. J Biological Chemistry 1993;268:23116-21.
- Johnson JA. Racial differences in lymphocyte beta-receptor sensitivity to propranolol. Life Sciences 1993;53:297-304.
- Kennedy JL, Petronis A, Gao J, et al. Genetic studies of DRD4 and clinical response to neuroleptic medications. Am J Hum Genet 1994;55:suppl:A190.
- Lefkowitz RJ, G-protein-coupled receptors: Turned on to ill effect. Nature 1993;365: 603-4.
- Propping P, Nothen M. Genetic variation of CNS receptors: A new perspective for pharmacogenetics. Pharmacogenetics 1995;5:318-25.
- Van Tol HHM, Wu CM, Guan H-C, et al. Multiple dopamine D4 receptor variants in the human population. Nature 1992;358:149-52.
|



