|
|
| What Happened to Tommy? |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, November 1998, 363-367
|
I started out my postgraduate medical training in pathology, and, perhaps for that reason, have always had a fondness for clinical pathological conferences. These exercises are perhaps the most classic and best teaching techniques in medicine. In such conferences, a clinician is provided with the medical history, physical examination, and laboratory test results and asked to arrive at a clinical diagnosis--which is then compared to the gold standard, the pathological diagnosis. In previous columns, I have used an analogous approach in presenting clinical psychopharmacology cases. The cases I have discussed in previous columns have typically dealt with patients who were taking multiple medications which interacted in complex ways. In this column, however, I present a much simpler case but one which nevertheless illustrates some important pharmacological principles.
THE CLINICAL DATA
Tommy was a 7-year-old boy with attention deficit disorder (ADD). He weighed 55 pounds (i.e., 25 kilograms). Tommy had been taken to a physician for treatment of his ADD. Methylphenidate was tried first, but produced no meaningful improvement. Tommy was next treated with imipramine, beginning with a dose of 50 mg/day. The dose was gradually titrated up to 200 mg/day over a period of 3 months. This titration was done on the basis of the usual clinical assessment of response (i.e., incomplete efficacy and good tolerability), which was done at approximately biweekly appointments.
In the spirit of a clinical psychopharmacologic conference (CPC), take a moment to reflect on what has been presented. Why review such a simple case? What’s the point? After you have formulated your opinion, read on.
One day at school, Tommy went to the water fountain to get a drink and collapsed. His teachers initiated cardiopulmonary resuscitation and called 911. Although Tommy’s heartbeat was re-established, he had fixed and dilated pupils and no spontaneous respiration. He was transported to a local emergency room (ER) where he was maintained on a ventilator while physicians worked to understand and treat the cause of his collapse.
The electrocardiogram (EKG) recorded during his ER stay showed ventricular tachycardia and fibrillation. He had one regular QT complex with an interval of 580 milliseconds (msecs) (the upper limit of normal for a child of 6 is 450 msecs). These findings are classic for tricyclic antidepressant (TCA)-induced cardiotoxicity. Therapeutic drug monitoring (TDM) in the ER revealed a total TCA plasma level of 1,000 ng/ml (imipramine plus desipramine). Parenthetically, TDM had never been done before during Tommy’s treatment. Shortly after these tests were obtained, Tommy had a recurrent bout of ventricular fibrillation and then a final cardiac arrest.
Again in the spirit of a CPC, reflect on the case and formulate your thoughts as to why Tommy died. What do you think his autopsy showed?
THE POSTMORTEM
The postmortem examination was performed 20 hours after Tommy was declared dead. It revealed no anatomical cause of death (i.e., no pulmonary embolus or other acute pulmonary disease, no anomaly of his intracardiac conduction system or coronary arteries, and no brainstem hemorrhage). His postmortem total TCA femoral blood level was 6,500 ng/ml (imipramine plus desipramine).
WHY DID TOMMY’S LEVELS GO UP AFTER DEATH?
While Tommy’s antemortem total TCA plasma level was 1,000 ng/ml, his postmortem TCA blood level was 6,500 ng/ml. This difference is due to the well-established phenomenon of postmortem re-equilibration between deep compartments (e.g., adipose tissue) and the central compartment (e.g., blood).1 Psychiatric drugs are highly lipophilic so that they can cross the blood-brain barrier and reach their site of action. If they did not, they would not have psychotropic effects. In addition, most but not all are highly protein-bound. Because of these two characteristics, most psychiatric drugs have a large apparent volume of distribution. In other words, they accumulate in disproportionately higher levels in tissue compartments than in plasma.
Whole blood has both a tissue component (i.e., red and white blood cells and platelets) as well as plasma water and plasma protein. When a whole blood sample is obtained, it is spun to separate the plasma (i.e., plasma water and plasma protein) from the tissue component.
The drug level is then measured only in the plasma. In fact, one can also measure only the "free" concentration in the plasma water if desired by separating the plasma water from the plasma protein. However, this approach is not typically used in routine clinical TDM.
When someone dies, he or she undergoes autolysis (i.e., their tissue breaks down). During this process, drug is released from the tissue compartments into the plasma. As a result, the concentration measured in the postmortem blood sample will be higher than the antemortem plasma level. That is why one should not jump to conclusions based solely on postmortem levels. However, some have tried to argue that this phenomenon completely invalidates postmortem levels. That is not true. Furthermore, we have cases like Tommy’s in which there are multiple factors supporting the unequivocal conclusion of iatrogenic poisoning.
SO WHAT HAPPENED?
Tommy’s case, while tragic, is one of the best but not the only example of iatrogenically induced fatal TCA poisoning. This conclusion is based on the following: 1) the unusually high dose of imipramine; 2) the high antemortem TCA plasma level; 3) the classic EKG findings of TCA poisoning; and 4) the absence of any other cause of death based on the autopsy.
This case illustrates two pharmacological principles. First, dosing rate and clearance are equally important to patient outcome because they determine the drug concentration. Second, drug concentration is important because it determines which site(s) of action will be engaged by the drug and to what extent.
As regular readers of this column know, there is a straightforward relationship between drug concentration, dose, and clearance as expressed by the following equation:
Concentration = Dosing rate / Clearance (Equation 1)
The reason that we have any "usual" doses is that human beings are more alike than different, because we share a common genetic code. Hence, we are more alike than different in our drug clearance. For that reason, a sizable percentage of the population will develop similar drug concentrations on a given dose of a given drug. That is the principal reason why we have a "usual" dose, but there is more.
Based on its structure, a drug has different affinities for different "targets" such as receptors, transporter proteins (i.e., uptake pumps), enzymes, and ion channels. These targets are the product of genes. Targets such as transporter proteins are therefore more alike than different in members of the same species since they share the same genetic code. That is the second reason for a "usual" dose.
The affinity of a drug for a specific target determines what concentration of that drug is needed to occupy that specific target to a specific degree. Stated another way, the drug’s concentration determines what target(s) will be occupied and to what extent in a given patient. This is why drug concentration is important.
A sign of pharmacological naïveté is the belief that dose is important but concentration is not. Dose is only important because it determines drug concentration and thus determines what target(s) will be engaged and to what extent. Almost every one knows that drugs have dose-response curves. However, everyone does not appreciate that these are actually concentration-response curves. Dose is simply a shorthand and imperfect way of expressing drug concentration.
Results in preclinical studies are often expressed in terms of dose for two reasons. First, it is a less time-consuming and less expensive approach than measuring drug concentration. Second, preclinical scientists use highly inbred strains of laboratory animals for their experiments. They do so because these animals are bred to be genetically quite similar. For that reason, there is a closer one-to-one relationship between dose and concentration in such animals. In other words, one goal of the inbreeding is to eliminate much of the variance in drug clearance between members of the same strain so that the clearance variable drops out of equation 1 and dose becomes equivalent to concentration.
As Lindsay DeVane has said: "A man is not a mouse."2 The genetic difference between mice and men, such as those coding for drug metabolizing enzymes, such as cytochrome P450 enzymes, in large measure are responsible for the differences in drug clearance between these species. Those differences, in turn, substantially contribute to the differences between mice and men in dose-concentration and hence dose-response curves. That is why predicting human doses from animal pharmacokinetic studies is fraught with difficulty. Moreover, humans avoid inbreeding; there is therefore much more variance between different members of our species than between members of the same strain of mice or rats used in preclinical scientific studies. That in turn results in much greater variance in dose-concentration and hence dose-response curves in humans than in research animals. That is the rationale behind TDM (i.e., to reduce at least some of this variance by being able to rationally adjust the dose to compensate for interindividual differences in drug clearance).
Returning to the case at hand, Tommy was not an unusual human being in terms of his drug clearance. Rather, he was on a dose that would produce toxic levels in most humans. When Tommy’s case occurred, the manufacturer and the Food and Drug Administration warned in the package insert for imipramine that: "A dose of 2.5 mg/kg/day should not be exceeded in childhood. ECG changes of unknown significance have been reported in pediatric patients with doses twice this amount."3 There have been formal studies which used doses of imipramine up to 5 mg/kg/day in children but only with appropriate safeguards (TDM and EKGs).4 In Tommy’s case, the physician used a dose of 8 mg/kg/day without any safeguards besides clinical assessment. The only humans who might need the dose Tommy was taking would be the relatively rare individual who is a genetically determined ultra-rapid CYP 2D6 metabolizer.
Since Tommy did not have unusually rapid metabolism, he developed an antemortem total plasma level of imipramine and desipramine of 1,000 ng/ml. As I discussed in my March 1998 column ("Marooned: Only One Choice"), tertiary amine tricyclic antidepressants (TCAs) such as imipramine have rather similar affinities for a number of different targets. For this reason, they have several different dose(concentration)-response curves that can be clinically meaningful (Figure 1). As I discussed in that earlier column, there is only a five- to ten-fold difference between the concentration of imipramine needed to block the uptake pumps for norepinephrine and serotonin (i.e., the putative mechanisms mediating its antidepressant activity) and the concentration needed to inhibit sodium fast channels. It is this latter mechanism that can cause drugs like imipramine to slow intracardiac conduction. When that conduction is slowed excessively, there can be escape ventricular beats and eventually fatal arrhythmias, as happened to Tommy.
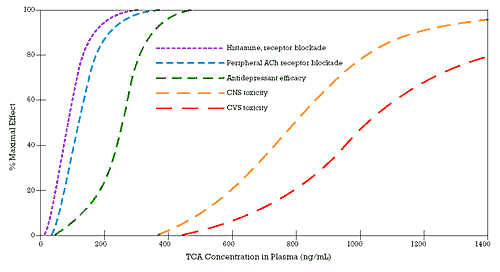 | | Figure 1 - Multiple Concentration: Response Curves of Tertiary TCAs |
AN ALL TOO COMMON ERROR
This case also illustrates the logical error that "more must be better." Insufficient dose is not the only nor even the most likely cause of inadequate drug efficacy. It is more likely that the patient has an illness that is fundamentally not responsive to the mechanism of action of the drug. A second explanation is that the drug level may be too high rather than too low, which is just the opposite of assuming the prescriber should "push the dose until you see something." That approach may be fine in rigorous controlled clinical trials, such as are done in a phase I research unit.
Consider the care taken in trying to determine the optimal dose in clinical drug development. First, dose finding studies in phase I are done only after there has been extensive experience with the drug at much higher doses in animals. In those animal studies, drug assays are now done to determine what the drug exposure was, given the fact that animals, particularly rats and mice, generally clear drugs considerably more efficiently than do humans. These days, it is also common for the human metabolism of the drug to have been studied in vitro using human CYP enzymes to better anticipate the dose needed in humans to achieve drug concentrations that have proven safe in animals. In the first human studies, single doses of the drug are given starting at a dose well below one which would be expected to produce any effect. The first goal is to establish the "no effect" dose. After that, single doses are escalated in separate groups of subjects with each escalation being based on a careful assessment of the safety and tolerance of the preceding dose. That assessment includes monitoring vital signs, hematological and chemistry laboratory tests, and EKG as well as evaluation of subjective tolerability. Other special tests may also be done depending on the nature of the drug.
Multiple dose exposure (e.g., repeated daily dosing) is done only after the safety and tolerability of single dose exposure has been determined. The multiple dose phase I studies are undertaken with the same "due diligence." Clinical trials to determine efficacy are undertaken only after the multiple dose, Phase I studies have determined a safe and well-tolerated dosing range. The maximum dose in the clinical trials for efficacy never exceeds the dosing range established by the phase I studies and is generally below the maximum dose tested in those earlier studies.
Once the drug is approved, clinicians may prescribe doses that exceed the recommended range, but, in doing so, they are engaging in research. This discussion is not meant to imply that clinicians should never exceed the recommended dosing range, but rather that they should do so with due caution and with the patient’s informed consent. There may well be a few patients who need higher doses due to the same genetic variability among humans mentioned earlier (e.g., the ultra-rapid CYP 2D6 metabolizers).
In fact, detecting such patients is one reason to employ TDM. If a patient does not benefit from a drug at its usually effective dose, it may be due to rapid clearance, which can be assessed by TDM. If the patient develops levels well below those usually expected on the usually effective dose, this would provide a rationale for cautiously employing higher doses with the caveat that the prescriber needs to be confident that the lower levels are not the result of noncompliance (see my May 1996 column "To Monitor or Not to Monitor?")
Human variance can also result in patients developing toxicity despite being on a dose which is well within the range that is usually safe and well tolerated. In the case of the TCAs, CYP 2D6 poor metabolizers can develop toxic levels on "usual" doses. Such poor metabolism can be due either to genetic deficiency in the enzyme or to the coadministration of a drug that produces substantial inhibition of this CYP enzyme (e.g., fluoxetine).
CYP 2D6 poor metabolism coupled with the narrow therapeutic index of TCAs is the reason why I have long advocated that TDM be employed once early in treatment (i.e., during the first week on a stable dose) as a standard of care. The rationale is to detect such slow metabolism so that an appropriate dose adjustment can be made.
NOT UNIQUE
Sadly, Tommy’s case is not the first instance of sudden death on a TCA due to substantially exceeding the recommended dosing range. Over 20 years ago, a case was reported in which a 14-year-old girl died suddenly and unexpectedly during treatment with imipramine at a dose of 14.5 mg/kg/day.5 Still, such cases are rare. More common are cases in which the patient was on a dose within the recommended range but nevertheless developed toxic levels. The problem in these cases is unusually slow metabolism either because of genetic deficiency in CYP 2D6 or because of the coadministration of drugs which substantially inhibit CYP2D6 (e.g., fluoxetine or paroxetine) as discussed in my September 1998 column "Sweetness"). I am aware of approximately 30 such cases of TCA-associated sudden death.6-8
Cases such as Tommy’s will become even rarer as the use of TCAs decline and the use of dose adjustment based on TDM becomes the accepted standard of care when prescribing these drugs. Nevertheless, Tommy’s case underscores the importance of several general pharmacological principles:
- Dose and clearance are equally important in determining drug concentration.
- Drug concentration determines the nature and magnitude of the drug’s effect.
- Some drugs have multiple different concentration-response curves due to their ability to affect multiple different sites of action.
- Dose cannot be escalated with immunity.
- Dose adjustment based solely on clinical assessment of response is not a foolproof way of avoiding serious toxicity.
- TDM can be a useful way of assessing the appropriateness of a specific dose of a specific drug for a specific patient.
- The concept of drug redistribution underlies the phenomenon of postmortem redistribution and can cause elevation in apparent plasma drug levels.
The most important and basic "take home" message of this column is that the warning, "too much of a good thing can be bad," is as applicable to medicine as it is to life in general.
References
- Prouty RW, Anderson WH, Postmortem redistribution of drugs. In: Jones GR, Singer PP, eds. Proceedings of the 24th International Meeting. International Association of Forensic Toxicologists. Edmunton, Alberta, Canada: University of Alberta Printing Services; 1988:1-21
- DeVane CL, Dr. DeVane replies. J Clin Psychiatry 1996;57(5):225-7
- Physicians’ Desk Reference, 2nd edition. Montvale, NJ: Medical Economics; 1995:578-90
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Safar KR, Klein DF, Gittleman-Klein R, Groof S, Imipramine side effects in children. Psychopharmacologia 1974;37:265-74
- Preskorn SH, Therapeutic drug monitoring of tricyclic antidepressants: A means of avoiding toxicity. In: Dahl S, Graham L, Eds. Clinical pharmacology in psychiatry: From molecular studies to clinical reality. Berlin: Springer-Verlag; 1989:237-43
- Preskorn SH, Sudden death and tricyclic antidepressants (TCAs): A rare adverse event linked to high TCA plasma levels. Nord J Psychiatry. 1993;47(suppl 30):49-55
|



