|
|
| Defining "Is" |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, July 1999, 224-228
|
The inspirations for this column often come from questions posed by colleagues around the country. One frequently asked question is whether differences in the in vitro potency of medications have any physiological/clinical implications. This question can take many forms such as:
- Is citalopram the most "selective" SSRI?
- Is sertraline an inhibitor of dopamine uptake?
- Is paroxetine a blocker of muscarinic acetylcholine receptors?
As President Clinton might say, the answer to these questions depends on the definition of "is."
In the first question, if "is" is limited to a consideration of the in vitro potency of citalopram for the serotonin versus the norepinephinre uptake pump, then the answer is "Yes." If "is" in this question takes into account the effects on other neural targets and the effects of the drug's metabolites, then the answer is "No." If "is" in the questions about sertraline and paroxetine refers to in vitro potency, then the answer is "Yes." If "is" refers to what happens in vivo under clinically relevant dosing of these two SSRIs, then the answer is probably "No."
Rather than playing semantic games with the answers, I will use this and several subsequent columns to expound on the following questions:
- What is the relationship between in vitro potency and in vivo effects?
- What does "selectivity" mean?
- How does "selectivity" relate to modern psychiatric drug development?
- What are the implications of such development for our understanding of psychiatric illnesses and their treatment?
This series of columns is based in part on material contained in two of my recent books. In this column, I will briefly review in vitro binding affinity and the concept of pharmacological selectivity. In the next column, I will discuss antidepressants that have sequential mechanisms of action and present the visual representation of the neural mechanisms of action of the various classes of antidepressants. The third column in this series will present a table summarizing the adverse effect profiles of prototypic antidepressants from each of the eight classes outlined in my 1998 column, "Marooned: Only One Choice."
The following equation is central to to any discussion of selectivity:
Effect = potency at x drug x biological
site of action level variance
In previous columns, we have focused on either drug level or biological variance as determinants of drug effects. In this series of columns, we will focus on potency at site of action.
From Chance to Planned Discovery
The modern era of antidepressants began with the chance discovery of monoamine oxidase inhibitors (MAOIs) (e.g., phenelzine, tranylcypromine) and tertiary amine tricyclic antidepressants (3°TCAs) (e.g., amitriptyline, imipramine). The discovery of these drugs began with failed attempts to develop treatments for tuberculosis and preanesthetic agents, respectively. Chance discovery of the first effective medications is not unique to psychopharmacology but has been the rule in virtually every therapeutic area.
The alternative to chance discovery is planned discovery. The latter requires some knowledge about the mechanism of action needed to produce the desired clinical effect. Therefore, drugs discovered by chance have historically served as the first step in discovering such mechanism(s) of action. The logic is as follows: If drug A produces a desired clinical effect, then drug A must affect a site of action X that mediates that effect. The isolation and identification of that mechanism permits the rational search for drugs that have the same mechanism. To improve on drugs discovered by chance, newer agents are synthesized so that they do not produce the undesired effects associated with the chance discovery, prototype drug. Such synthesis requires the development of structure-activity relationships (SARs) based on hypotheses generated from studying the effects of the chance discovery agents. The goal is to produce new molecular entities (NMEs) that have both high potency and selectivity for a specific mechanisms) of action.
As explained in an earlier column, "Marooned: Only One Choice," amitriptyline -- the prototypic 3°TCA -- served as the blueprint for the development of six new classes of antidepressants. The strategy was to develop NMEs that mimicked only the pharmacology of amitriptyline needed to produce antidepressant efficacy while avoiding the actions that accounted for its toxicity and tolerability problems.
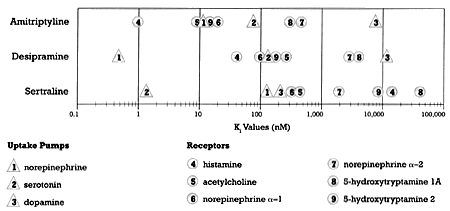 | | Figure 1 - Relative potency for different sites of action of three different types of antidepressants |
Figure 1 shows the binding affinity of amitriptyline, desipramine, and sertraline for a variety of specific neural sites of action.5,6 The most potent action of amitriptyline is blockade of the histamine-1 receptor. However, as explained in the "Marooned" column, amitriptyline is essentially a "cocktail" or a fixed combination of single mechanism of action drugs. As the dose of amitriptyline is increased, its concentration increases, which in turn sequentially engages sites of actions that have a lower affinity for the drug, but are still clinically meaningful (Figure 1). The concentration of amitriptyline needed to treat clinical depression is consistent with its ability to block the neural uptake pumps for both norepinephrine and serotonin.7 At those concentrations, amitriptyline also blocks histamine, muscarinic acetylcholine, and alpha-1 adrenergic receptors. That is the reason amitriptyline produces adverse effects before it produces clinical benefit and why it produces so many different types of effects and can be used for so many different clinical purposes. The "take home message" is: The binding affinities of a drug determine the concentration of the drug needed to engage its targets to a physiologically meaningful degree.
Binding affinity is the first term in equation 1. Concentration is the second term. The concentration in turn determines the dose needed in the patient relative to his or her clearance of the drug as explained by equation 2:
Concentration = dosing rate / clearance
In essence, randomized clinical trials are population pharmacokinetic studies in which the goal is to determine the "usual" dosing rate needed in the "usual" patient (i.e., "usual" clearance) to achieve a concentration of the drug that engages its "therapeutic" mechanism of action to the degree necessary to produce the desired clinical effect.
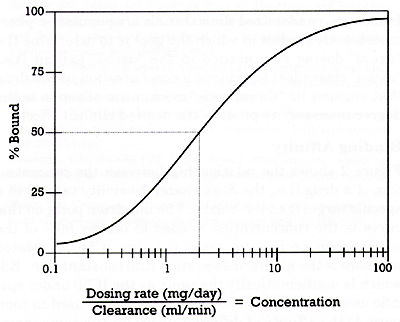 | | Figure 2 - Relationship between receptor occupancy (% bound) and drug concentration (i.e., dosing rate / clearance) |
Binding Affinity
Figure 2 shows the relationship between the concentration of a drug (i.e., the X-axis) and its ability to bind to a specific target (i.e., the Y-axis). The inflection point on this curve is the concentration needed to occupy 50% of the receptors in an in vitro assay (i.e., the IC50). A related measure is the kinetic dissociation rate constant (i.e., Kd) which is mathematically the same as the IC50 under specific assay conditions. IC50s and Kds can be used to compare: 1) the affinity of different drugs for the same target (i.e., the serotonin uptake pump) or 2) the affinities of the same drug for different targets (i.e., the serotonin versus the norepinephrine uptake pump).
Based on such data, structure-activity relationships (SARs) can be deduced to guide the synthesis of compounds with specific binding characteristics. It was just such an approach that led to the development of the SSRIs and other new antidepressants.1,2 This knowledge is also central to answering the questions posed at the beginning of this column. Parenthetically, the absolute values for IC50s and Kds for a drug can vary from one laboratory to another depending on the conditions used in the in vitro assay. In a later column, I will describe the degree to which such results can vary and how to avoid misinterpreting the data.
Thus, amitriptyline and the other antidepressants that were discovered by chance served as the blueprints needed to produce SARs that in turn could be used to guide the development of new antidepressants. The power of this approach is evident from the fact that there are now eight classes of antidepressants as classified by mechanism of action.3
In addition to amitriptyline, Figure 1 shows the binding affinity profile of two other classes of antidepressants, norepinephrine selective reuptake inhibitors (NSRIs), as represented by desipramine, and serotonin selective reuptake inhibitors (SSRIs), as represented by sertraline. As can readily be seen, these two types of antidepressants differ from amitriptyline in several important ways. First, their most potent binding affinity is for a site of action that mediates antidepressant response. Second, there is a substantial gap between their first and second targets. It should be noted that these two drugs differ from each other in terms of their most potent site of action. Desipramine and sertraline bind most strongly to the norepinephrine and the serotonin uptake pumps, respectively. This difference in apparent mechanism of action mediating antidepressant efficacy is consistent with the fact that 50% or more of patients who do not benefit from a NSRI or from an SSRI will respond when switched to the other class.
Selectivity
The discussion above sets the stage for understanding the concept of pharmacological selectivity or constraint. Recall that measurements of binding affinity such as IC50s are concentration-dependent terms. Recall also that
Concentration = dosing rate / clearance
In a given individual, the above equation reduces to:
Concentration = dosing rate
as long as the drug has linear pharmacokinetics in that specific patient (i.e., clearance is independent of dose over the dosing range of interest). Note that most drugs have linear pharmacokinetics. For that reason, the gap between a drug's first and second site of action indicates how much the dose has to be increased, if at all, in that individual to affect the second site of action -- the wider the separation, the larger the dose increase needed to affect the next target. Viewed from another perspective, a drug with a wide gap can achieve a concentration that achieves substantial occupancy and hence effect on the first target while at the same time achieving minimal occupancy and hence minimal effect on any other targets. This is the definition of drug selectivity. A parallel concept is pharmacological constraint, which means the drug's effects are limited to those mediated by the drug's most potent site of action.
As a general rule of pharmacology, a drug is said to be selective if there is a 10-fold (i.e., one order of magnitude) difference between its binding affinity for its first versus its second target. The greater the gap, the more selective the drug is. These principles are illustrated in Figure 3.
| Drug A with Three-Fold Difference in Binding Affinity for Receptor X versus Receptor Y | 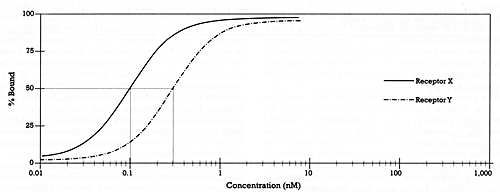 | | Drug B with 1 Order of Magnitude Difference in Binding Affinity for Receptor X versus Receptor Y | 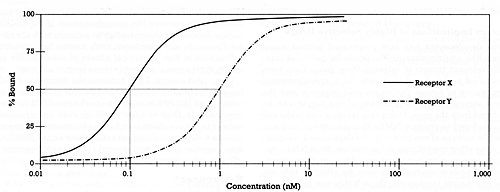 | | Drug C with 2 Orders of Magnitude Difference in Binding Affinity for Receptor X versus Receptor Y | 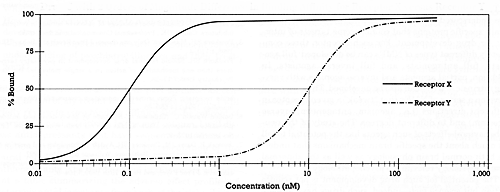 | | | Figure 3 - The relationship between drug concentration and occupancy (% bound) of two different targets (receptors X and Y) for three different drugs |
The top graph in Figure 3 illustrates a drug with a 3-fold difference in binding affinity for target X versus Y. This drug will always have a greater effect on X than Y. Nevertheless, substantial occupancy of X (i.e., 50% or more) cannot be achieved with such a drug without also producing some occupancy of Y. The middle graph in Figure 3 shows a drug with a 10-fold (i.e., one order of magnitude) difference in binding affinity for X versus Y. With this drug, virtually complete occupancy of X can be achieved at a concentration that produces only minimal occupancy of Y. The bottom graph shows a drug with a 100fold (i.e., two orders of magnitude) difference in affinity for X versus Y. In this case, no detectable occupancy of Y is achieved at a concentration that produces full occupancy of X. Once that degree of separation has been achieved, further increases in selectivity (i.e., three or four orders of magnitude) may be of interest to the researcher but are of doubtful clinical consequence -- a comment that may help to put into proper perspective marketing claims about how "selective" a drug is.
As a general rule, approximately 70%-80% occupancy of a site of action appears to be needed for most psychiatric drugs to achieve a clinically meaningful effect.2,4,7 Increases in occupancy above 80% are often associated with more of an increase in adverse effects than in efficacy. This general rule is consistent with the fact that all the fixed dose studies with single mechanism of action drugs (e.g., SSRIs) show a flat dose-response curve with regard to efficacy but an ascending dose-response curve with regard to adverse effects above their usually effective minimum antidepressant dose.8
Future Implications of Highly Selective Drugs
Drug development has now progressed beyond using drugs like amitriptyline as blueprints for potential clinically relevant targets of interest. Once genes have been identified and isolated that code for a specific neural mechanism of action, the amino acid sequence and the three dimensional configuration of the target can be deduced from the gene. Using this information, SARs can be developed to produce NMEs that selectively bind to a specific subtype of receptor (e.g., the dopamine-4 receptor) or some other type of neural mechanism. Such NMEs can then serve as new investigational drugs and simultaneously as probes into brain function. By affecting only one discrete neural mechanism, these NMEs can have highly discrete neurobehavioral consequences.
The enormous implications of such drug development are underscored by estimates that there are 5,000 or more brain specific proteins that could become targets of interest for drug development. For each receptor, three completely different types of NMEs can be developed: full agonists, full antagonists, and full inverse agonists. In addition, partial agonists and inverse agonists with varying intrinsic activity can also be developed. Thus, we could develop a series of NMEs that "tweak" a given receptor in different directions (i.e., agonism, antagonism, inverse agonism) and to different degrees. The study of the neurobehavioral effects of such agents has the potential to tell us much about the specific brain mechanisms that underlie both normal brain physiology as well as the abnormal brain physiology relevant to psychopathology.
The potential for such drug development is not science fiction but science fact -- indeed, such drugs have already been developed and tested. A major challenge now is to define their human behavioral pharmacology. The old ways of characterizing the neuropsychopharmacology of such drugs (i.e., preclinical animal models of psychiatric illnesses and randomized clinical trials in patients meeting current criteria for psychiatric syndromes) may well be as much of a hindrance as an aid. Even testing in normal humans is likely to be inadequate to characterize the pharmacology of such drugs in specific forms of psychopathology. This is because of the third variable in equation 1 (i.e., biological variance among different individuals). That variance can include the pathophysiology that is mediating the patient's symptoms. In other words, there may be an interaction between the neurobehavioral effects of highly selective NMEs and the pathophysiology of the patient's illness.
For these reasons, we advocate more extensive neurobehavioral testing of truly novel NMEs in individuals who are mildly symptomatic with the target illness during late Phase I, either before or concurrent with larger scale Phase II testing. We believe that such testing can substantially enhance the characterization of the NME's human behavioral pharmacology in patients with the target illness. In our experience, such testing has revealed differences in the behavioral effects of novel NMEs in mildly symptomatic patients versus normal subjects.
In my next column, I will continue the discussion of differences in binding affinities and in vivo effects as they relate to all the SSRIs as well as to the non-SSRI antidepressants. In that column, I will provide an expanded answer to the questions posed at the beginning of this column as well as explaining why venlafaxine has ascending dose-response curves with regard to both antidepressant efficacy and increases in blood pressure.
References
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Preskorn SH, Should rational drug development in psychiatry target more than one mechanism of action in a single molecule?. Intl Rev Psychiatry. 1995;7:17-28
- Cusack B, Nelson A, Richelson E, Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559-565
- Bolden-Watson C, Richelson E, Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
|



