|
|
| Why Did Terry Fall Off the Dose-Response Curve? |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, January 1996, 39-43
|
In this column, I begin a series addressing a frequent, vexing problem in clinical psychopharmacology: why do some patients not respond optimally to the usually effective dose of a medication? Patients may fall off the standard dose-response curve in several ways: the patient who does not improve on the usually optimal dose (i.e., an insensitive benefit outlier) or who does not tolerate the usually optimal dose (i.e., a sensitive tolerance outlier). There is also the inverse of each of these categories: the patient who responds at a dose much smaller than is usually necessary (i.e., the sensitive benefit outlier) or who appears to tolerate a much higher dose than usual (i.e., an insensitive tolerance outlier). While the latter two types of patient typically cause less clinical concern, they are of equal scientific interest. Before considering why Terry fell off the dose-response curve, I will discuss what a dose-response curve is, how it is determined, and why it is clinically important.
WHAT IS A DOSE-RESPONSE CURVE?
The determination of a drug's dose-response curve(s) is fundamental to basic and clinical pharmacology. As illustrated in Figure 1, the magnitude or nature of the response to a drug is determined by the dose of the drug. More specifically, the response is a function of the drug concentration at the site of action as expressed in equation 1:
Clinical response = Potency for site of Action
x Drug concentration at site of action
Dose, however, is often used as a simple, shorthand surrogate for concentration. This substitution enables us to avoid having to measure the drug concentration, which can be technically complicated, particularly early in a drug's development, and is not necessary if there is a highly predictable relationship between dose and concentration. There is also the issue of where to measure the drug concentration. Obviously, the biologically meaningful concentration is that at the site of action -- but that is virtually never measured due to technical difficulties. Instead, the drug concentration in plasma is typically used because it is more readily sampled and because it often correlates well with the tissue concentration of the drug in the organ of interest (e.g., brain, heart).
WHAT IS THE PURPOSE OF ESTABLISHING THE USUAL DOSE-RESPONSE CURVE?
The goal is to determine whether the clinical response (the variable reflected on the Y-axis of Figure 1) is a function of the dose (the variable expressed on the X-axis of Figure 1). In any such curve, the variable on the Y-axis is the dependent variable and the variable on the X-axis is the independent variable. One way to statistically test whether there is a functional relationship between these two variables is to do correlational statistics. In this statistical approach, the goal is to determine whether a statistically significant amount of the variability in the dependent variable is determined by the variability in the X-axis (e.g., drug dose or concentration). Since we are talking about variability, it is obvious that we are not talking about the response in a single patient at a single time but rather the variance in the response of different patients receiving the same dose at the same time under the same conditions or the variance in the response of the same patient but at different times (e.g., response to the same drug at different times for recurrent episodes of an illness). The first is the situation most often studied because the issue is how most patients with the same illness will respond to a given dose of the same drug. Such data will inform the physician of what dose should be used when treating most patients.
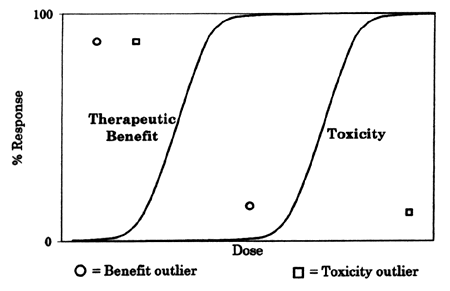 | | FIGURE 1 - Response to drug X as a function of dose |
HOW IS THE USUAL DOSE-RESPONSE CURVE DETERMINED AND WHAT IS THE SIGNIFICANCE OF A FIXED DOSE VERSUS A FLEXIBLE DOSE DESIGN?
To illustrate how this type of population dose-response curve is established, we will do an experiment in which we will treat 100 patients with a drug. For our example, the drug will be an antidepressant such as a selective serotonin reuptake inhibitor (SSRI). This class of drugs was chosen for our initial example because they appear to have only one mechanism of action relevant to their central nervous system (CNS) effects (e.g., the inhibition of the neuronal uptake pump for serotonin). Moreover, that mechanism is the same for every member of this class. This fact is why the CNS pharmacology of these drugs is so similar. (There are, however, clinically important differences between members of this class with regard to other effects, in particular the inhibition of specific P450 enzymes which appears to mediate the potential for altering the clearance rates of substrates normally metabolized by these enzymes -- but that is a topic for another column.) The reason we want to use a drug with only one mechanism of action for our initial example is so that only one mechanism will determine the clinical outcome.
For our experiment, we randomly assign our patients to 1 of 10 predetermined dosing groups ranging from a low to a high dose based on the results of preclinical studies. The purpose of these preclinical studies was to determine the potency of the drug for affecting the site of action that we think is going to be responsible for its desired effects in a specific disease (e.g., major depression). This design is called a fixed dose study because the dose is predetermined. In such a design, the dose is the independent variable.
An alternative design is a flexible dose study in which the clinician can titrate the dose based on clinical response. In this design, there is a complicated relationship between the response and the dose because neither is truly independent of the other. The initial response is determined by the starting dose but then the dose becomes a function of the patient's response. Moreover, the dose is also a function of the idiosyncrasies of the clinician, who may be a proponent of the "more is always better" philosophy or who may a priori believe that a higher dose of a drug is necessary to obtain the desired response. Since the "more is always better" philosophy is rampant in American psychiatry, flexible dose design studies almost always overshoot the usually effective minimum dose of a drug. For this reason, ascending dose design studies generally determine the maximally tolerated dose rather than the usually effective dose. The reason is because the upward titration in such studies is most often limited only by dose dependent adverse effects due to the drug's excessive action on the site(s) mediating the drug's effect.
The fixed dose design avoids all these complications. Using this approach, we randomly assign patients to one of the 10 different dose levels in a double-blind fashion (i.e., neither the doctor nor the patient knows which dose level the patient is receiving). We then follow the patient during the treatment trial and determine one of three outcomes: the drug works, the drug does not work, or the drug has to be stopped because of nuisance and/or serious adverse effects.
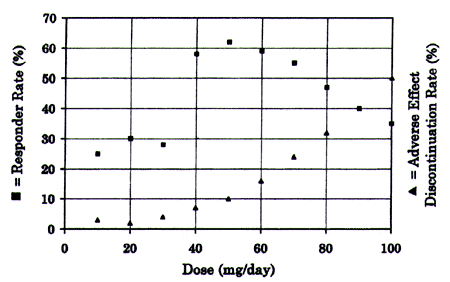 | | FIGURE 2 - Dose Dependent curves for responder rate and adverse effect discontinuation rate |
Figure 2 shows the typical outcome of such a study. In constructing this figure, I assumed a placebo response rate of 25% and a maximal drug response rate of 65%, assumptions that describe the usual result in clinical trials of marketed antidepressants. In our experiment, the lowest doses of our drug produced neither benefit nor adverse effects in most patients. At intermediate higher doses, an ever increasing percentage of patients respond to the drug until a plateau is reached in the response rate. Our study found that the usually effective minimum dose was 40 mg/day. Doses above 40 mg/day did not result in a greater percentage of responders. In fact, the percentage of responders at higher doses actually began to decease as the rate of early discontinuation due to adverse effects increased. At least one reason why the percentage of responders decreases at higher doses is that the percentage of early dropouts due to dose-dependent adverse effects is greater than any increase in the percentage of patients who require a higher dose to respond. Early dropouts are patients who have to stop treatment so early that they did not have long enough exposure to the drug to receive any therapeutic benefit. The failure of these patients to respond is counted as a treatment failure because they were assigned to treatment at that specific dose and did not benefit, even though they may have responded had they been assigned to a lower dose. The point of this study is not to get all patients well but rather to determine the dose of the medication that works best for the largest percentage of patients. The usually effective minimum dose is the dose that overall treats the largest percentage of patients, taking into account those who must drop out due to adverse effects before efficacy can be reasonably expected. There is no reason to increase the dose above the usually effective minimum dose in the "usual" patient.
WHAT ABOUT VARIABILITY AMONG PATIENTS?
Of course, there is substantial variability in how different patients respond to the same dose of the same medication. That is why there are standard deviation bars around the mean response value. However, the usual dose-response curve (Figure 1) only reflects the average response in the overall population being treated in the study. Patients who are at the extremes of the standard deviation in terms of response to that dose are the outliers I mentioned at the beginning of this column. These outliers can deviate from the average by being either more or less sensitive to either the beneficial or the adverse effects of the drug, thus making up the four categories described at the beginning of the column. Since the clinician is interested in maximizing the response in all patients rather than just the "usual" patient, the question becomes why do some patients deviate from the usual and what, if anything, can be done to adjust the dose to allow these patients, as well as the "usual" patient, to be treated optimally with the drug. I will focus more on the issue of outliers in my next column.
WHY DOES THE DROPOUT RATE FOR DRUGS WITH A SINGLE MECHANISM OF ACTION INCREASE WITH DOSE ESCALATION?
As a general rule of thumb, excessive engagement of any site of action will cause adverse effects mediated by that site of action without further increasing the magnitude of the desired effect. This appears to happen because the body can no longer compensate for the effect of the drug on that site of action and consequently adverse effects result. Take the SSIs as an example. All members of this class of antidepressants have a flat dose-antidepressant response curve. The usually effective minimum dose of each of these drugs produces 70%-80% inhibition of the serotonin uptake pump using the platelet as a surrogate marker for what is happening in the brain. Although these drugs have a flat dose-antidepressant curve, they have an ascending dose-adverse effects curve, meaning that the incidence and severity of adverse effects of these drugs increase with increasing doses even though antidepressant efficacy does not. These adverse effects include nausea, diarrhea, anxiety, and insomnia. These adverse effects appear to be produced by excessive inhibition of the same site of action that mediates the desired antidepressant response, the serotonin uptake pump.
WHAT ABOUT DRUGS THAT HAVE MORE THAN ONE MECHANISM OF ACTION?
Such drugs typically produce more than one physiological effect. They therefore produce more than one clinical response, typically in a dose-dependent (i.e., concentration-dependent) manner as dictated by the drug's binding affinities for these different sites of action. The effects produced by the drug at different concentrations may be desirable or undesirable, depending on their magnitude and/or the clinical situation. Obviously, any marketed drug must produce some effects that are considered clinically desirable at doses lower than those that cause adverse effects. If that were not the case, the drug would never have been marketed because it would not be clinically useful and hence would not be commercially viable.
Tricyclic antidepressants (TCAs) and low potency phenothiazines (e.g., thioridazine) are excellent examples of drugs that affect multiple sites of action over their clinically relevant dosing range. The most potent action of many tertiary amine TCAs (e.g., amitriptyline) is the blockade of histamine receptors. For this reason, low doses of these drugs can produce sedation and interact pharmacodynamically with other sedative hypnotic agents (e.g., alcohol) to potentiate their sedative effects. This sedative action may be desirable or undesirable depending on the patient and the clinical situation. At therapeutic doses (i.e., concentrations), TCAs block or slow both norepinephrine and serotonin uptake pumps which are believed to mediate their antidepressant efficacy. At concentrations above those needed to treat major depression, these drugs block fast sodium channels which produces slowing of intracardiac conduction. Even the latter effect can be beneficial in the right degree and in the right clinical situation. For example, TCAs can suppress arrythmias. However, this action can also result in serious intracardiac conduction delays and cause arrythmias when the effect is excessive.
In the case of drugs such as TCAs or phenothiazines, if the dose is increased sufficiently above the plateau for therapeutic benefit, the drug is likely to produce adverse effects that range from nuisance to treatment-limiting to serious and potentially life-threatening. One major goal of the clinical trial phase of a drug's development is to determine the drug's dose-response curve in terms of both efficacy and toxicity. The difference between what is routinely a therapeutic versus a toxic dose is the drug's therapeutic index.
HOW DOES THE RESPONSE IN THE INDIVIDUAL PATIENT RELATE TO THE USUAL DOSE-RESPONSE CURVE?
The usual dose-response curve represents the usual response in the general population. For the individual patient, we must modify equation 1 as follows:
Clinical response = Potency for site of Action
x Drug concentration at site of action
x Underlying biology of patient
The first variable in both equations 1 and 2 is the drug's pharmacodynamic profile or what the drug does to the body. The drug must affect a site of action physiologically capable of mediating the desired clinical response. It does so by binding to this site of action and either activating or inhibiting it. The result of the interaction of the drug with the site of action (i.e., activation or inhibition) is termed the drug's mechanism of action (e.g., inhibition of the neuronal uptake pump for a specific neurotransmitter or the activation or blockade of a specific neuroreceptor).
The second variable in both equations 1 and 2 is the pharmacokinetic profile of the drug in that patient (ie., what that patient's body does to that drug in terms of absorption, distribution, and metabolism of elimination). That variable determines what concentration of the drug will be achieved in the body of that patient on a given dose of that specific drug. To produce a clinically meaningful response, the drug must reach a critical concentration in the body to engage its mechanism of action to a sufficient extent to produce a physiologically meaningful effect. That is the domain of pharmacokinetics and is heavily dependent on that patient's rate of clearance of that drug, which in turn is heavily dependent on the cytochrome P450 enzymes that were the subject of my last column.
The third variable in equation 2 is underlying biological variance in the patient, which shifts the patient's individual dose-response curve from the "usual" dose-response curve either to the left (i.e., more sensitive) or to the right (i.e., less sensitive). That underlying biological variance may be produced by genetic, environmental, or disease factors or the interactions of more than one of these factors. The study of these variables could be termed the "biopsychosocial" approach to drug response (Figure 3).
 | Age
(puberty,
menopause)
Time of day
Food effect
Concomitant
drug effect
Disease | Genetics:
pharmacokinetics
pharmacodynamics
Gender
Environment
(shared) | Disease
Concomitant
drug effect
Body size
Environment
(nonshared) |  | | | | | FIGURE 3 - Reasons for pharmacological variability |
HOW HAVE PHYSICIANS HISTORICALLY TRIED TO ADAPT THE USUAL DOSE-RESPONSE CURVE TO A SPECIFIC PATIENT?
For a long time physicians have used clinically recognizable differences between patients to make dose adjustments for the "not usual" patient. The admonition, "start low and go slow," is just that sort of dose adjustment. It is commonly recommended for elderly patients because of concern that they may be more sensitive than the "usual" patient to the adverse effects of a drug at a given dose. For the same reason, physicians also frequently reduce the "usual" starting dose of a medication in the medically ill, particularly those with cardiac, liver, and renal disease. These practices may be scientifically supported by evidence showing that such patients clear drugs from the body at a much slower rate than the "usual" patient and will thus develop higher drug concentrations on the same dose and/or that patients in these "not usual" categories are indeed more likely to develop significant adverse effects on the "usual" dose than will happen in the general population (i.e., the "usual" patient).
Physicians also tailor the dose for the patient who outwardly appears "usual" but does not respond like the "usual" patient by titrating the dose based on a clinical assessment of the patient's response. Here is a typical scenario. The physician starts at the "usual" dose. If the patient does not experience the desired beneficial response and has no apparent adverse effects, the physician increases the dose until either the desired response occurs or adverse effects become dose-limiting. In the latter case, the physician may stop the drug or add another drug. The second course of action can begin a cycle of polypharmacy for the patient, which is a topic for another column. If the patient does not tolerate the "usual" dose from the beginning, then the above process goes in the opposite direction.
WHAT ARE THE LIMITATIONS OF TRYING TO TITRATE THE DOSE BASED ON CLINICAL ASSESSMENT OF RESPONSE? ARE THERE OTHER ALTERNATIVES?
Although time honored, this titration approach has several shortcomings. It is slow, error prone, and costly. The dose adjustment is not made until the patient has either failed to respond adequately or has had an adverse effect. Since many psychotropic medications take time to work, the patient must often remain ill for several weeks before a decision can be made that the dose is inappropriate. Moreover, the dose-dependent (i.e., concentration-dependent) adverse effects of many psychotropic medications can mimic the illness being treated (e.g., TCAs can cause dysphoria, irritability, decreased concentration, or fatigue), making it difficult to know whether to go up or down on the dose in the event of a suboptimal response. The typical reaction to an insufficient response is to increase the dose, but a dose increase in such cases may cause a worsening of the patient's condition.
Rather than relying on such a trial and error method to determine the optimal dose, it would obviously be desirable to be able to detect the "unusual" patient before starting treatment so that appropriate dose adjustment could be made at the beginning. That dream is becoming a reality thanks to the field of pharmacogenetics, which is expanding our knowledge of why Terry falls off the usual dose-response curve. I will discuss this topic in my next column.
Suggested Readings
- Melmon KL, Morelli HF, Hoffman BB, Nierenberg DW, eds. Clinical pharmacology: Basic principles in therapeutics. New York: McGraw-Hill,1992.
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr. Principles and practice of psychopharmacotherapy. Baltimore: Williams & Wilkins, 1993.
|



