|
|
| To Monitor or Not to Monitor? |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, May 1996, 172-175
|
That is the question. When is it nobler and better patient care to use therapeutic drug monitoring (TDM) as opposed to the time honored approach of dose titration based on clinical assessment of response? In this column, I continue the discussion I initiated in the last one.
Primary Goal: Improved Safety
A misconception on the part of many psychiatrists is that the primary goal of TDM is improved efficacy. It is not. The primary goal is improved safety.
In medicine, the guiding principle has traditionally been: "First, do no harm." As medicine has set its sights on tackling more serious illnesses, that principle remains fundamental to good patient care -- but its application is perhaps a bit more difficult. The reason is that any potent medication can cause adverse effects. A literal interpretation of the "do no harm" principle could mean that many of the potent and effective weapons of modern medicine against disease should not be used. Such a position would be untenable in an era when we seek to kill cancerous cells to save the body, crack chests to repair damaged hearts, and use medicine capable of causing pseudomembranous colitis to cure life-threatening bacterial infections. Instead, the principle has come to be interpreted using a more complex equation that balances the risk posed by the untreated disease, the likelihood and magnitude of the benefit that can reasonably be expected from the treatment, and the likelihood, potential seriousness, and reversibility of adverse effects that might occur from the treatment.
Psychiatrists, fortunately, have not escaped the necessity of having to apply such equations to their care of patients. I use the word "fortunately" because the need for such equations results from having potent and effective treatments. When treatments are not potent or likely to produce benefit, the likelihood that they will directly cause harm is also modest. In order for a drug to be approved, the likelihood of the drug producing benefit must outweigh the likelihood that it will cause harm and this judgment must be made within the context of the disease being treated. If the likelihood of a fatal outcome due to a disease (e.g., an aggressive malignancy) is high and imminent without treatment, there is a higher tolerance for the treatment itself having the potential to cause toxicity. The more benign and self-limited the disease (e.g., the common cold), the less tolerance there is for the treatment being likely to cause toxicity.
Like malignancy, psychiatric illnesses such as acute psychotic disorders and severe depressive episodes can have a high and imminent risk of fatality. Even milder forms of these illnesses can greatly impair functioning and cause appreciable pain and suffering for patients and their loved ones. These risks must be balanced against the risk of treatment. For this reason, medications with a known risk of causing toxicity are used when necessary to treat patients suffering from these disorders.
What Features Dictate TDM?
Some notable examples of medications with a known risk of causing toxicity include: bupropion, clozapine, carbamezapine, lithium, low potency antipsychotics, and tricyclic antidepressants. This list includes a diverse group of psychiatric medications: antidepressants, antipsychotics, mood stabilizers. Why then are they grouped together? All these medications have well documented dose-dependent risks of toxicity (Table 1) -- in other words, their risk of toxicity increases predictably with dose increases. However, these drugs can be used clinically because the risk of toxicity is acceptable if the dose is in the range that is typically therapeutic. This fact strongly suggests that the drug has different mechanisms of action at these different doses (i.e., concentrations) which are responsible for these different clinical consequences (i.e., efficacy versus toxicity). This fact has been particularly well established for tricyclic antidepressants (TCAs): TCAs affect the uptake pumps for serotonin and norepinephrine at concentrations produced by doses that are generally therapeutic; however, they affect sodium fast channels at concentrations typically produced by doses that cause cardiac and central nervous system toxicity.
| Table 1 - Examples of psychiatric medications with dose dependent toxicity | | Drug | Toxicity | Bupropion
Clozapine
Carbamezapine
Lithium
Low potency antipsychotics
Tricyclic antidepressants | seizures
seizures
delirium
obtundation
cardiotoxicity, seizures
cardiotoxicity, seizures |
Dose dependent toxicity is one of the features of a drug that predicts the benefit of using TDM to help guide dose adjustment. Other features are listed in Table 2. If a drug affects multiple sites of action over its clinically relevant dosing range, then it may affect sites at one concentration that are irrelevant to the desired effect but capable of causing adverse effects (e.g., as with TCAs). If there is large interindividual variability in the clearance of the drug, then patients who clear the drug slowly may develop toxic concentrations on conventional doses. The toxicity may develop insidiously or present suddenly. Since the onset of efficacy is delayed for many psychiatric medications, the ability to titrate the dose based on response is impaired, and the clinician does not know whether the problem is an inadequate dose or inadequate duration of treatment. If a drug has one or more of the features listed in Table 2, then dose adjustment based on TDM may be a significant improvement over dose titration based on clinical assessment of response.
| Table 2 - Features predicting the usefulness of therapeutic drug monitoring | • Multiple mechanisms of action
• Dose dependent, serious toxicity
• Larger interindividual variability in metabolism
• Insidious onset of toxicity
• Delayed onset of efficacy |
How Would TDM Help?
In essence, establishing a therapeutic range for a drug is simply a refinement of establishing the therapeutic dosing range. The therapeutic dosing range determines the typical concentrations that will occur in most patients in a population whereas TDM determines the concentration that occurs in a specific patient. TDM allows the physician to detect and adjust for differences in the elimination rate of the drug in different patients. As I explained in the last column, that difference may be due to genetic, disease, or environmental factors.
Figure 1 illustrates a drug that has a genetically determined trimodal population curve with regard to drug concentration achieved on a given dose, with the low dose represented by dose A, medium dose by dose B, and high dose by dose C. In this example, the drug has a minimum plasma drug concentration threshold for efficacy and a maximum threshold above which the risk of toxicity increases disproportionally to any increase in efficacy. Due to the trimodal distribution, there is no single dose which places all patients within the range that achieves maximum efficacy with minimum risk of toxicity, which, parenthetically, is the definition of the therapeutic range. Obviously, this definition involves weighing the likelihood of further efficacy against the risk of serious adverse consequences.
| Shift in the drug concentration population frequency curve as a function of increasing dose in a polymorphic population | 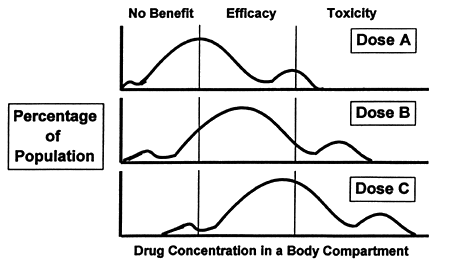 | | Figure 1 - Distribution of plasma levels of a drug in a heterogeneous population as a factor of dose |
Figure 1 is not fantasy. It is based on the plasma levels of imipramine that are achieved in a population of physically healthy depressed patients on the same dose of this drug. The patients who develop high levels on this dose are individuals who are functionally deficient in the cytochrome P450 enzyme which is principally responsible for metabolizing TCAs, CYP 2D6. The large, middle group is composed of individuals who have at least one normal gene for CYP 2D6. The small group who develop unusually low levels for the dose given are individuals with a relatively rare variant of the gene which makes them unusually efficient metabolizers via CYP 2D6.
Without measuring the functional activity of the enzyme, the clinician would have no way of knowing whether the patient is a slow, normal, or ultra rapid metabolizer via CYP 2D6. One way to make that determination is by measuring the levels of the drug that the patient develops on the dose being taken. That approach would then allow the physician to adjust the dose to compensate for interindividual differences in elimination rate.
Another approach to the use of this drug would be to simply mandate a conservative dose (i.e., dose A in Figure 1) to ensure that the smallest possible percentage of patients have a chance of developing a toxic concentration. The downside to this approach is that many patients will develop levels that are below the minimal effective threshold and thus will not benefit from treatment. Alternatively, the physician might take an aggressive approach to ensure that as many patients as possible have a chance of responding to the drug by using a dose that will produce plasma levels above the minimum effective threshold in most patients (i.e., dose C in Figure 1). The problem with this approach is that a sizable percentage will develop levels that put them at risk for experiencing toxicity. The intermediate strategy would be to split the difference and use dose B.
Some may say that the minimum dose and maximum dose approaches mentioned above are inappropriate and never used and that instead physicians simply titrate the dose based on their assessment of the patient's response. Those individuals would be wrong. Take bupropion as an example. That drug is explicitly labeled with a maximum recommended daily dose. That approach is based on the fact that bupropion has a dose dependent risk of seizures and that the risk in a general population at doses of 450 mg/day or less is considered acceptable relative to the benefits of the drug in treating a serious illness, major depression. Nonetheless, this approach is analogous to dose A in Figure 1. It sets a dose threshold to ensure that the percentage of the population at risk is acceptable. It does not address the risk in those patients who, on 450 mg/day, are functionally on a much higher dose due to slow clearance. At the same time, a maximum recommended dose ensures that patients who rapidly clear the drug will functionally be on a dose that is below the usually effective dose for most patients.
Generally, the toxic threshold is not based on the most scientifically rigorous data which would come from a formally designed study to establish such a threshold. The reason is ethics. A rigorous approach would by design place some patients at increased risk for developing toxicity. Two designs for such a study could be used. The first would be to treat a large population with a high dose (i.e., dose C in Figure 1), which should place a sizable percentage of patients at risk for developing toxicity, and then determining the threshold above which the risk substantially increased. The other approach would be to randomly assign two groups to different concentration ranges and then determine whether there is a significant difference in the rate of toxicity between the two groups.
Instead of such prospective "toxicity" studies, the toxicity threshold is estimated by extrapolation from population-dose data or from case report data in which plasma drug levels were obtained in patients who experienced toxicity on conventional doses. In the population-dose approach, the incidence of serious toxicity is plotted as a function of dose and the same is done for plasma drug levels. These data typically come from two different studies: the adverse effect data as a function of dose from large scale efficacy studies and the plasma levels as a function of dose from smaller pharmacokinetic studies. The data are then examined for natural breaks (as illustrated in Figure 1) and to determine whether the percentage of the population under these two curves are similar for both plots. If the incidence of toxicity rises as a function of dose, this observation strongly implicates a concentration-dependent effect. If the percentage of the curves under the two plots are similar, this fact further supports such a relationship and implies that concentration above a specific point increases the risk of this adverse effect. This relationship can be further tested by obtaining plasma drug levels in patients who inadvertently experience the toxicity in question either during clinical trials with the drug or after it has been marketed. In addition to finding that the incidence of the toxicity is dose-dependent, a relationship between drug concentration and a toxic effect is also indicated when the incidence of the toxic effect is temporally linked to drug administration (e.g., occurs at time of peak plasma drug concentrations) and to dose increases (e.g., occurs within the time that a new steady-state level should be achieved after a dose increase) and when patients who slowly clear the drug or rapidly absorb it are at increased risk. Many of these observations, in addition to dose dependency, have been made for the drugs listed in Table 1 in relationship to their specific toxic effect.
In this column, we have discussed the primary goal of TDM -- to increase the safety of treatment by avoiding high concentrations that are associated with an increased risk of toxicity. The ultimate goal of TDM, and of good patient care in general, is to reduce the variance in response and make it more predictable. However, TDM will not eliminate all variance because there are other variables (e.g., organ impairment due to disease or age, genetically-determined differences in end organ responsivity) that can shift the concentration-response relationships (as I discussed in my columns in the September 1995 and January 1996 issues).
In future columns, I will explore those interindividual issues further and discuss advances that are being made in detecting and adjusting for such differences to further reduce variance in treatment response. In the next column, I will continue the discussion of TDM and address secondary but clearly important additional goals of TDM -- which include increasing efficacy by establishing the minimum threshold below which efficacy is appreciably reduced and serving as a means of assessing compliance. I will specifically address the problems that are encountered when trying to establish the minimum effective threshold due to the large "signal-to-noise" ratio in efficacy studies in psychiatry. I will also discuss when physicians should or could use TDM (i.e., when is TDM an option and when is it a necessity?)
Suggested Readings
- Mormon K, Hoffman B, Nierenberg D. Introduction to clinical pharmacology. In: Melmon KL, Morrelli HF, Hoffman BB, Nierenberg DW, eds. Clinical pharmacology: Basic principles in therapeutics. New York: McGraw-Hill; 1992:7-19.
- Davidson J. Seizures and bupropion: A review. J Clin Pechiatry 1989;50:266-61.
- Preskorn S. Should bupropion dosage be adjusted based upon therapeutic drug monitoring? Psychopharmacol Bull 1991;27:637-43.
- Preskorn S, Burke M, Fast G. Therapeutic drug monitoring: Principles and practices. In: Dunner D, ed. The Psychiatric Clinics of North America. Philadelphia: WB Saunders; 1993:611-46
|



