|
|
| Finding the Signal through the Noise: The Use of Surrogate Markers |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, March 1999, 104-109
|
Research is like trying to detect a homing beacon (i.e., the signal) in dense fog (i.e., the noise). Obviously, the larger the signal-to-noise ratio, the greater the likelihood that the research will succeed. Thus, the successful researcher is the one who can increase the sensitivity of his or her tools for picking up the signal, while simultaneously dampening down the interfering background noise.
Locking onto the homing beacon is a vexing problem for the psychiatric researcher and, in turn, for practicing psychiatrists and their patients. The problem is the low signal-to- noise ratio typical of most psychiatric research. It hampers the testing of virtually all investigational psychiatric medications. In this column, I will discuss this subject and its relationship to the use of surrogate markers to:
- better define the optimal dose (i.e., concentration) of psychiatric medications, and
- elucidate clinically important biological variance in the site of action of a drug (i.e., the first and third variables in equation 1).
(Equation 1)
Effect = site of x drug x biological
action level variance
This column is taken in part from a chapter I wrote on the problem of establishing dose-response relationships for psychiatric medications for a book published under the auspices of the European Commission on Scientific and Technical Research.1
While the topics covered in this chapter are relevant to all medications (psychiatric and nonpsychiatric), I will use the selective serotonin reuptake inhibitors (SSRIs) as an example because they were the first rationally designed class of psychiatric medications intended to have a single and specific mechanism of action, indirect serotonin agonism via the inhibition of the neuronal uptake pump for this neurotransmitter.2,3 The statements in this column are applicable to any of the five SSRIs marketed in the United States; however, I will use sertraline as the prototype because of the extent of data relevant to this topic on this SSRI.
The Problem of a Low Signal-to-Noise Ratio in Clinical Psychopharmacology Trials
In essence, clinical trials are population pharmacokinetic studies that attempt to determine what dose will achieve a drug concentration that will engage the desired mechanism of action to a necessary and sufficient degree (i.e., above the minimum threshold for efficacy) in the greatest percentage of patients enrolled in the trial without overengaging that mechanism of action and thus causing adverse effects (i.e., above the maximum threshold for tolerability). The fixed dose study is the most common research approach used to make this determination.
In a fixed dose study, patients are randomly assigned in a double-blind fashion to receive one of several predetermined doses of the investigational medication or placebo. This research approach to dosing is quite different from clinical practice in which physicians typically take into account readily discernible biological variance in their patients to select what they hope will be an optimal dose for the particular patient (e.g., the adage of "start low and go slow" when dosing frail, geriatric patients). In contrast to this individuation of dose in clinical practice, the dose for each patient in a fixed trial is determined by the proverbial "flip of a coin." Moreover, most studies do not employ a "go slow" approach to dose titration for the higher dose groups. Instead, patients either start on the higher doses immediately or are rapidly titrated to their predetermined target dose. If patients cannot tolerate their assigned dose, they drop out. Often, patients drop out before they could reasonably have been expected to have responded to the drug. Thus, these patients end up as both early dropouts and as non-responders in the traditional last-observation-carried-forward data analysis approach for the "intent-to-treat" population
While this approach may seem odd to the clinician, remember the goal is to determine the optimal dose of a new drug. The researcher does not know the answer and is using this design to seek it (i.e., trying to find the homing beacon through the fog of ignorance). The practitioner should realize that the results from such trials determine the dosing recommendations recorded in the package insert to assist clinicians in optimally dosing their patients.
A major confounding factor in this endeavor is the poor signal-to-noise ratio caused by the inability to select study participants who are uniquely responsive to the mechanism of action of the drug. Response in such trials is typically defined as a 50% reduction in symptom severity between the beginning and the end of the clinical trial (i.e., usually 6 weeks). Using this definition, the most common finding in such a trial is that one-third of patients on placebo respond versus two-thirds on the antidepressant--which leaves one third who do not respond.4 Thus, the "signal" in clinical trials is the one patient out of three who specifically benefits because of antidepressant treatment (i.e., overall drug response minus placebo response). The "noise" is the placebo response plus the nonresponse (i.e., two patients out of three). Thus, the "signal-to noise ratio" in antidepressant clinical trials is 1:2, which is poor.
Parenthetically, the placebo condition in such a trial is often incorrectly equated with "no treatment." However, patients on placebo in such clinical trials receive a substantial amount of treatment (i.e., more than 10 hours over 6 weeks) in the form of good clinical management (i.e., education about the illness and supportive psychotherapy) from well-trained research staff. The reader interested in further discussion of this topic is referred to an earlier column.4
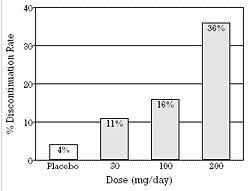 | Figure 1 - Discontinuation Rate Due to Adverse Events as a Function of Predetermined, Fixed Doses of Sertraline.
Based on data from Fabre et al.5 Similar data are available for fluoxetine (Wernicke et al.6,7) and paroxetine (Dunner and Dunbar8). |
Nonresponders in a clinical trial may occur for a variety of reasons. In a fixed dose study, they may have been assigned to a dose that is ineffective either because it results in concentrations below a minimum threshold needed for efficacy or because it results in concentrations above a maximum tolerability threshold (Figure 1). Alternatively, they may have a form of the disease that is not responsive to the mechanism of action of the drug and would therefore not respond regardless of what drug concentration is achieved.
The inability to screen out placebo responders and nonresponders from clinical trials has multiple consequences. First, it increases the number of subjects who must be enrolled in a clinical trial to determine a group difference between patients on the antidepressant versus those on placebo (i.e., "power analysis"). Second, it severely compromises the ability to establish a classic ascending dose-antidepressant response relationship. Third, it makes it virtually impossible to determine a concentration-antidepressant response relationship.
A further complication in antidepressant research is the strong possibility that antidepressant response is a step rather than a graduated function (i.e., an "all or none" phenomenon which requires reaching a necessary and sufficient threshold for efficacy). Supporting this conclusion is the fact that 80% of patients who have a 50% reduction in symptom severity (i.e., the definition of response) actually experience a full remission (i.e., become asymptomatic). The fact that fixed dose studies with most of the newer antidepressants, including all the SSRIs, have found a flat dose-antidepressant response is also consistent with the conclusion that antidepressant response follows a step function (i.e., no average gain in response above the lowest dose which is effective) (Figure 2). The finding of a flat dose-antidepressant response curve does not mean that every patient needs the same dose but rather that an equal number of patients in the intent-to-treat population will respond to each dose using the last-observation-carried-forward analysis.
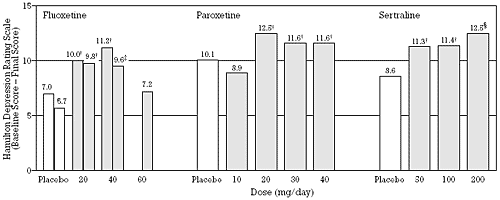 | Figure 2 - Antidepressant Efficacy as a Function of Predetermined, Fixed Doses of Three SSRIs.*
Based on data from Wernicke et al.,6,7 Dunner and Dunbar,8 and Fabre et al.5 | *Comparable data from fixed dose studies have not been published for citalopram or fluvoxamine
†p < 0.05
‡p < 0.01
§p < 0.001 compared to placebo |
While the antidepressant response to the SSRIs is flat, all of these drugs produce an ascending dose-response curve with regard to dropouts due to tolerability problems (Figure 1). The occurrence of an ascending early dropout rate at higher doses means that fewer patients who could potentially respond remain in the study on these higher doses. In fact, the approach of using a last-observation-carried-forward analysis for the "intent-to-treat" population means that a greater percentage of the patients remaining on higher doses have to respond to produce a response rate equal to that seen on lower doses.
For all these reasons, it is difficult, if not impossible, to establish a classic, ascending dose-antidepressant response curve with antidepressants, particularly those like the SSRIs that were designed for safety and tolerability reasons to have a single mechanism of action over their clinically relevant dosing range.
Use of Surrogate Markers in Clinical Trials
Surrogate markers for drug effect are a widely accepted tool in drug development. In some instances, surrogates (e.g., receptor occupancy in the brain as measured by positron emission tomography) are used during Phase I testing to determine a dose that will achieve a concentration that is likely to be clinically effective in Phase II trials. In other instances, drug approval is based on effects on surrogate markers rather than the target disease. For example, lipid lowering drugs were initially approved on the basis of their ability to reduce the levels of cholesterol and specific forms of triglycerides. These drugs were developed as a way of reducing the risk of atherosclerosis. If the drugs had been required to prove initially that they reduce atherosclerosis, they might not have been developed, given the length of time it would have been necessary to give the drugs to demonstrate a clinically meaningful reduction in the development of atherosclerosis. On the other hand, there was reasonable but not indisputable evidence that elevated levels of cholesterol and specific forms of triglycerides were etiologically important risk factors for the development of the disease. Thus, lowering these levels became an accepted surrogate marker for the effect of these drugs on the target disease.
There are several obstacles to the use of surrogate markers in psychiatry. First and foremost, the pathophysiology and pathoetiology of most psychiatric illnesses has not been established. Thus, a surrogate cannot be selected on the basis of its established relevance to the illness. Second, most psychiatric illnesses are syndromes and thus are likely be found to be more than one illness when understood at the level of pathophysiology and pathoetiology. This is certainly likely to be the case with major depression, based on the available clinical and pharmacological evidence. The inescapable conclusion that comes from examining the results of genetic studies, biological marker studies, and clinical trials in patients with major depression is that this syndrome is unlikely to be due to a single pathophysiology or pathoetiology. In fact, such biological heterogeneity likely accounts for many of the "signal-to-noise" problems in clinical trials of antidepressants discussed above.
Despite these limitations, there is considerable evidence to suggest that platelet serotonin uptake inhibition can be a useful marker for the clinically relevant pharmacology of SSRIs. The rationale behind this statement is reviewed next.
The Central Serotonin Hypothesis of Clinical Depression
In the 1960s and 1970s, studies of the pharmacology of tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) led to the hypothesis that central serotonin agonism was a viable means of producing an antidepressant response. The active transport of serotonin across the cell membrane has been recognized for at least 40 years (see Sneddon 19739 for a review). Shortly thereafter, it was recognized that drugs that inhibited this transporter protein were indirect central serotonin agonists. These observations led several pharmaceutical companies in the 1970s and 1980s to search for drugs that inhibited this transporter protein without affecting other neuroreceptors. This effort resulted in the development of a number of the newer antidepressants including citalopram, fluoxetine, fluvoxamine, paroxetine, sertraline, and venlafaxine. The first five comprise the class of antidepressants known as SSRIs,3 while venlafaxine is an example of a serotonin and norepinephrine reuptake inhibitor.10
The success of the SSRI class is reflected in the fact that, in 1998, one out of two prescriptions for an antidepressant in the United States was for a member of this class.11 The widespread acceptance of SSRIs is due to their efficacy coupled with excellent safety and good tolerability in comparison to older medications. At the same time, their success gives further credence to the idea that a disturbance in central serotonin function is causally connected to the occurrence of clinical depression.
Physiological Evidence for the Use of the Platelet Serotonin Uptake Pump as a Surrogate Marker in Psychiatry
Studying the direct effect of a drug on mechanisms in the brain is technically difficult for a number of reasons, including but not limited to the following. The brain is encased in a bony vault. Its output is complex. A direct causal relationship between a drug’s action and an effect is difficult to establish because there is the possibility of multiple intervening synaptic contacts which can modify the expression of the effect.
The platelet has been employed for a number of years as a model for the central serotonin neuron.12 The primary structure of the human platelet serotonin uptake site is identical to that of the brain serotonin transporter13 and the accumulation of serotonin in both tissues proceeds by analogous mechanisms that share a large number of identical characteristics.14 The drug-induced inhibition of the uptake pump is consistent with the drug-induced inhibition of neuronal firing in the following way. By blocking the serotonin uptake pump, the drug increases serotonin availability to presynaptic autoreceptors, which in turn inhibit serotonin neuronal firings.15
This phenomenon provides a heuristic explanation for the delay between the onset of serotonin uptake inhibition and antidepressant efficacy that is consistent with the central serotonin hypothesis of clinical depression. While the first effect caused by serotonin uptake inhibition is a decrease in serotonin neuronal firing, over time, a down-regulation of the serotonin presynaptic autoreceptors occurs, which leads to a return to the baseline level of serotonin neuronal firing in the continued presence of the serotonin uptake inhibitor. Under these conditions, the availability of serotonin to postsynaptic neurons increases, which leads to subsequent downregulation of specific postsynaptic neurons. The time course of these events parallels the time course needed for antidepressant response. That these events are relevant to the antidepressant response is further supported by the observation that acute but reversible depletion of serotonin in the brain can precipitate a recurrence of depressive symptoms in patients who have been successfully treated with antidepressants that inhibit serotonin uptake.16
Clinical Evidence for the Use of the Platelet Serotonin Uptake Pump as a Surrogate Marker in Psychiatry
Prospective, random assignment, double-blind, fixed dose, placebo-controlled clinical trials have demonstrated that SSRIs have a usually effective minimum dose, while higher doses on average produce no greater percentage of responders (Figure 2).5-8 In vivo studies in humans that examine platelet serotonin uptake provide a heuristic explanation for the flat dose-antidepressant response relationship.17-21 Platelet studies suggest that the usually effective, minimum dose of each SSRI achieves a comparable degree of serotonin uptake inhibition that is associated with good antidepressant efficacy and tolerability and that a greater degree of uptake inhibition is not on average associated with an increase in efficacy because of an increased dropout rate due to increased problems with tolerability (Figure 1).
This minimum threshold can be estimated from studies in which inhibition of platelet 5-HT uptake is determined relevant to dose or plasma drug concentration. For example, inhibition of platelet 5-HT uptake was studied in 30 patients dosed with 5, 25, or 50 mg. citalopram for 4 weeks.17 Serum obtained weekly from each patient was incubated with platelets obtained from a healthy unmedicated subject; active uptake of 1 µM 5-HT was measured over 2 minutes. Inhibition was calculated by reference to the mean uptake rate in a total of 29 samples collected before treatment. A very high correlation (r = 0.96) was found between uptake inhibition and serum citalopram. At plasma levels associated with 40 mg/day dosing (i.e., its usually effective minimum dose based on its fixed dose acute efficacy trials), inhibition was 60%; at plasma levels associated with 20 mg/day dosing, inhibition was 50%. Despite these robust findings with the surrogate marker, there were no significant linear correlations between clinical improvement and either serum concentration of citalopram or inhibition of 5-HT uptake due to the previously mentioned problems with signal-to-noise in such studies.
Inhibition of platelet 5-HT uptake was studied in four subjects receiving 30 mg fluoxetine for 7 days followed by 20 mg for 23 days.18 By the end of the 30-day dosing period, 80% inhibition of active uptake (100 µM 5-HT measured over 10 min.) was apparent.
The inhibitory effect of fluvoxamine on platelet 5-HT uptake was studied in 7 (of an initial 17) patients who remained in a trial of 100 mg fluvoxamine for at least 12 weeks.19 Platelet uptake (during 2 min. exposure to 0.25-4 µM 5-HT) averaged 53% less than in patients receiving lithium at a dose adjusted to maintain a fixed plasma level. In 5 fluvoxamine patients who continued for an additional 12 weeks, inhibition reached 64% relative to inhibition in patients treated with lithium. Uptake inhibition at 12 weeks correlated well (r = 0.94) with plasma fluoxetine level. Plasma fluvoxamine levels were lower than expected at an effective dose: 53% inhibition was noted at 51.3 ng/ml and 64% inhibition at 68.3 ng/ml, whereas the plasma levels expected at the minimum effective dose are closer to 100 ng/ml.3 In the same investigation, 200 ng/ml fluvoxamine was added to platelet-rich plasma from healthy unmedicated subjects; inhibition of uptake under the same assay conditions was 85%. Overall, 70% would be a reasonable estimate of the extent of 5-HT uptake inhibition to be expected at the usually effective minimum dose of fluvoxamine.
The effect of paroxetine upon whole blood 5-HT and platelet 5-HT uptake was assessed in patients completing a 6-week trial of a 30 mg dose.20 Only 12 of the 15 patients demonstrated a decrease in whole blood serotonin, but those 12, as well as 2 of the 3 showing an increase, averaged 76% inhibition of uptake (over a 2 min. period at an unspecified 5-HT concentration); the third patient’s blood was not assayed. If one adjusts downward linearly to the usually effective minimum dose, one might anticipate 60% inhibition at a dose of 20 mg/day paroxetine. The insensitivity of whole blood 5-HT as a measure of uptake inhibition may be reflected in the finding that there was no significant correlation between the change in whole blood 5-HT during paroxetine treatment and either Hamilton scores or the blood level of paroxetine.
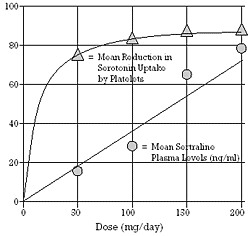 | Figure 3 - Relationship between daily dose of sertraline, mean plasma levels of sertraline and mean reduction in serotonin uptake by platelets after 14 days of drug administration at one of four fixed doses.
Based on data from Preskorn and Harvey.24 |
To assess the effect of sertraline on 5-HT uptake, normal male volunteers were dosed for a period of 2 weeks.21 Platelet uptake (during 2 min. exposure to 0.25-4 µM 5-HT) averaged 76% less on 50 mg/day of sertraline. This study provided data on uptake inhibition over the full range of efficacious doses, as shown in Figure 3. Even though the dose increased linearly, platelet uptake inhibition was near plateau levels at the minimum effective dose of 50 mg/day.
All the evidence described above is consistent with the concept that the antidepressant effect of SSRIs behaves like a step function rather than a continuous graduated function. In other words, a minimum threshold level of serotonin uptake inhibition is required for antidepressant efficacy (i.e., approximately 60%-80%) while inhibition above that level produces more of an increase in the discontinuation rate due to adverse effects than an increase in antidepressant efficacy.
Effect of Biological Variability on Platelet 5-HT Uptake
Within the last 10 years, the protein sequence of the serotonin membrane transporter has been determined.22 After that was accomplished, the issue of genetic variability in the 5-HT uptake transporter, which could in turn affect platelet 5-HT uptake, could be addressed. Research to date has found only minor degrees of variance in the sequence of the transporter itself across populations studied but has revealed common allelic variants in the promoter region of the gene which lead to more (allele l) or less (allele s) expression of the transporter itself.23 This finding is analogous to the genetic polymorphism in cytochrome P450 enzymes, such as CYP 2D6, discussed in earlier columns.24
Differences in frequencies of the allelic variants of the promoter region of the 5HT transporter gene predict differences in genotypes across populations: about 6% of African Americans, 16% of European Americans and 64% of Japanese have the s/s genotype.25 In vitro expression of the variants reflects two-fold differences in mRNA and 5-HT uptake across genotypes.23 Examination of post-mortem human brain reflects differences of a similar magnitude in uptake sites and transporter mRNA in relevant brain areas.26 Antidepressant response also varies with genotype, and the s/s genotype has been associated with a less robust response to fluvoxamine monotherapy.27
The effect of this biological variability in platelet 5-HT uptake has not been compared across cultures and most data have been reported by investigators at Caucasian/ European sites. Nevertheless, the platelet model has been used by Japanese to compare uptake inhibition with paroxetine binding to the transporter28 and to predict duration of in vivo inhibition from in vitro inhibition rate constants (Ki’s).29
Conclusion
Surrogate markers, such as platelet serotonin uptake inhibition, can be important tools in better understanding the clinically relevant pharmacology of new psychiatric medications. These tools will become even more important in the near future for two reasons. First, technology is improving our ability to design drugs to selectively fit a wide variety of specific targets such as uptake pumps, receptors, ion channels, and second messenger systems. Second, the completion of the human genome project will provide a rich and even bewildering array of potential targets for drug discovery, particularly in psychiatry, given the high percentage of the human genome that codes solely for brain specific proteins. Third, genetic research on psychiatric disease can narrow down the targets of interest by pointing to those that are likely to be patho-physiologically and pathoetiologically related to the disease process. Such research may well undercover mutation of a normal "wide type" receptor that underlies the expression of the disease. In fact, a different drug structure may be needed to fit that mutant than for the "wild type" version of the target. Even now, these principles are beginning to have an impact on the practice of psychiatry. By understanding them, practitioners can better conceptualize the treatment they provide for their patients.
References
- Preskorn SH, Recent dose-effect studies regarding antidepressants. In: Balant LP, Benitez J, Dahl SG, Gram LF, Pinder RM, Potter WZ, eds. European Cooperation in the Field of Scientific and Technical Research. Belguim: European Commission; 1998:45-61
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Preskorn SH, A dangerous idea. J Pract Psychiatry Behav Health 1996;2:231-4
- Fabre LF, Abuzzahab FS, Amin M, et al, Sertraline safety and efficacy in major depression: a double-blind fixed-dose comparison with placebo. Biol Psychiatry. 1995;38:592-602
- Wernicke JF, Dunlop SR, Dornseif BE, Bosomworth JC, Humbert M, Low dose fluoxetine therapy for depression. Psychopharmacol Bull. 1988;24:183-188
- Wernicke JF, Dunlop SR, Dornseif BE, Zerbe RL, Fixed dose fluoxetine therapy for depression. Psychopharmacol Bull. 1987;23:164-168
- Dunner DL, Dunbar GC, Optimal dose regimen for paroxetine. J Clin Psychiatry. 1992;53(suppl 2):21-26
- Sneddon JM, Blood platelets as a model for monoamine-containing neurons. Prog Neurobiol 1973;1:151-98
- Harvey AT, Preskorn SH, Mechanism of action of venlafaxine in normal male volunteers (Abstract). Clin Pharmacol Ther 1997;61:175
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Pletscher A, Platelets as models: Use and limitations. Experientia 1988;44:152-5
- Lesch K-P, Wolozin BL, Murphy DL, Riederer P, Primary structure of the human platelet serotonin uptake site: Identity with the brain serotonin transporter. J Neurochem 1993;60:2319-22
- Stahl SM, The human platelet. Arch Gen Psychiatry 1977;34:509-516
- Chaput Y, de Montigny C, Blier P, Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments: An in vivo electrophysiologic study in the rat. Neuropsychopharmacology 1991;5:219-29
- Miller HL, Delgado PL, Salomon RM, Licinio J, Barr LC, Charney DS, Acute tryptophan depletion: A method of studying antidepressant action. J Clin Psychiatry 1992;53(suppl 10):28-35
- Bjerkenstedt L, Flyckt L, Overo KF, Lingjaerde O, Relationship between clinical effects, serum drug concentration and serotonin uptake inhibition in depressed patients treated with citalopram. A double-blind comparison of three dose levels. Eur J Clin Pharmacol. 1985;28:553-557
- Lemberger L, Rowe H, Carmichael R, et al, Fluoxetine, a selective serotonin uptake inhibitor. Clin Pharmacol Ther 1978;23:421-9
- Wood K, Swade C, Abou-Saleh M, Milln P, Coppen A, Drug plasma levels and platelet 5-HT uptake inhibition during long-term treatment with fluvoxamine or lithium in patients with affective disorders. Br J Clin Pharmacol. 1983;15(suppl 3):365S-368S
- Marsden CA, Tyrer P, Casey P, Seivewright N, Changes in human whole blood 5-hydroxytryptamine (5-HT) and platelet 5-HT uptake during treatment with paroxetine, a selective 5-HT uptake inhibitor. J Psychopharmacol. 1987;1:244-250
- Preskorn SH, Harvey A, Biochemical and clinical dose-response curves with sertaline. Clin Pharmacol Ther. 1996; 59:180
- Ramamoorthy S, Bauman AL, Moore KR, et al, Antidepressant- and cocaine-sensitive human serotonin transporter: Molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci 1993; 90:2542-6
- Lesch K-P, Mossner R, Genetically driven variation in serotonin uptake: Is there a link to affective spectrum neurodevelopmental and neurodegenerative disorders?. Biol Psychiatry 1998;44:179-92
- Preskorn SH, Sweetness!. J Prac Psych and Behav Hlth 1998;4:304-8
- Gelernter J, Kranzler H, Cubells JF, Serotonin transporter protein (SLC6A4) allele and haptotype frequencies and linkage disequilibria in African- and European-American and Japanese populations and in alcohol-dependent subjects. Hum Genet 1997;101:243-6
- Little KY, McLaughlin DP, Zhang L, et al, Cocaine, ethanol, and genotype effects on human midbrain serotonin transporter binding sites and mRNA levels. Am J Psychiatry 1998;155:207-13
- Smeraldi E, Zanardi R, Benedetti F, Di Bella D, Rez J, Catalano M, Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Molecular Psychiatry 1998;3:508-11
- Nankai M, Yamada S, Yoshimoto S, et al, Platelet 3H-paroxetine binding in control subjects and depressed patients: Relationship to serotonin uptake and age. Psychiatry Res 1994;51:147-55
- Ishigooka J, Kasahara T, Nagata E, Murasaki M, Miura S, Effects of washing procedure on platelets pretreated with serotonin uptake inhibitors in vitro: Low Ki values predict long-lasting inhibition of serotonin uptake in vivo. Nihon Shinkei Seishin Yakurigaku Zasshi 1998;1:19-21
|



