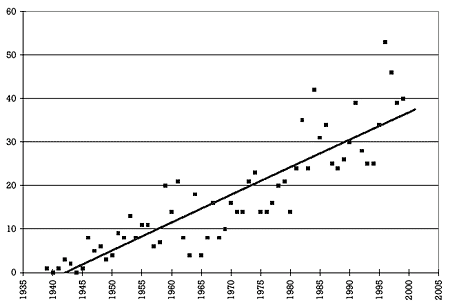
Figure 1. Number of drugs per year approved by the FDA for marketing in the United States 6,7
| Preskorn.com | Printed from: http://www.preskorn.com/columns/0201.html |
| Drug Approvals and Withdrawals Over the Last 60 Years |
|
|
|
SHELDON H. PRESKORN, MD |
|
Journal of Psychiatric Practice, January 2002, 41-50 |
The overarching theme of the current series of columns is drug development in psychiatry and the implications of the human genome project. Past columns have pointed out how pharmacogenetics and pharmacogenomics have accelerated drug development by identifying both desired targets and targets to be avoided. Scientists in drug discovery can use this information to design molecules with novel efficacy and improved safety and tolerability. Research in molecular biology and more traditional medicinal chemistry has also led to the development of "high throughput" screening technology, which allows drug discoverers to screen hundreds of thousands of molecular entities against all known human regulatory proteins of interest to develop more refined structure-activity relationships to guide drug synthesis.
With regard to improved safety, an earlier column in this series pointed out that certain widely used antidepressants such as fluoxetine and paroxetine would not be likely even to make it out of drug discovery today given our improved ability to design drugs to avoid unintended inhibition of cytochrome P450 enzymes.1 In terms of novel efficacy, earlier columns have pointed out the number of brain-specific regulatory proteins that have been identified as a result of the human genome project.2,3 Once identified and characterized, these regulatory proteins can become targets for developing drugs that affect specific central nervous system (CNS) functions. Such drug development has the potential to vastly increase the number of CNS drugs brought to market as well as the number of new indications for drugs. To put these advances in perspective, this column will review the number of drugs approved and withdrawn per year in the United States during the second half of the 20th century. The black box warning recently given to nefazodone due to drug-induced liver failure will also be discussed along with the potential role of pharmacogenomics research in salvaging such drugs for the majority of patients who could benefit from the drug without risk of this rare serious adverse event.
This column will point out limitations of the drug development and approval process. However, these comments require a context in which to be understood. In this regard, the words of Winston Churchill come to mind when he commented that democracy was a terrible form of government except when compared to all other forms of government. The drug development process in the United States and the other G7 countries is a business. That has consequences. Perhaps, the most important is that it has resulted in tremendous breakthroughs in the ability to treat and cure many, many diseases. That would likely not be the case if there was no incentive to risk the capital needed to do such groundbreaking work. Consider that few, if any drugs, have been developed by any government.
IMPLICATIONS OF DRUG APPROVALS AND WITHDRAWALS
The number of drugs approved and withdrawn is relevant to an issue that has been addressed periodically in this column, drug-drug interactions (DDIs) and their implications for the practitioner and the health care system in general (see www.preskorn.com for a list of relevant columns and publications). As the number of available prescription and over-the-counter (OTC) drugs and herbal products increases, the number of potential types of DDIs increases as a factorial of the number of marketed drugs that can be used in combination. Thus, the number of new drugs entering the market per year puts a strain on prescribers’ ability to stay current concerning the pharmacology of the individual drug and its potential DDIs. This issue is particularly vexing because DDIs occur across therapeutic classes. The prescriber needs to know not only about drugs approved in his or her therapeutic specialty but also about drugs used in all specialties, since another practitioner may prescribe a new drug that could adversely interact with one or more drugs in the regimen the patient is currently taking. This issue was highlighted in an earlier column that focused on a case in which four drugs from four different therapeutic classes prescribed by four different practitioners all interacted in a way that mimicked a worsening of one of the patient’s medical problems.4 That column and others made the point that DDIs can present in a myriad of masked ways, making them difficult to detect in clinical practice and, ironically, frequently leading to the use of more drugs to treat the signs or symptoms produced by the DDI.
The number of drugs withdrawn from the U.S. market underscores some of the limitations of the drug development process. Many of these drugs were withdrawn because of the risk of DDIs that were not detected during their development. Others were withdrawn because of quite rare but serious adverse events. Such events are most likely to be due to an interaction between the drug and some unique biology of the patient. Thus they are most likely to be understood within the context of the following equation:
Effect = affinity for + drug + biological
site of action concentration variance
(Equation 1)
The third variable refers to biological variance among humans that can either quantitatively or qualitatively alter the usual response to a given dose of a specific drug. Quantitative means that the response is the same but the magnitude is different. In other words, the dose-response curve in a specific individual has been shifted to either the left (i.e., less responsive) or to the right (i.e., more responsive) relative to the dose-response curve in the general population. Qualitative means the funda-mental nature of the effect is different. The major causes of biological variance among individuals include age, dis-ease, genetics, and internal environment (principally, the presence of drugs and other ingested substances). All of these variables have played a role in the withdrawal of drugs from the market over the last 37 years (the period for which these data are available).
A BRIEF QUIZ
Before proceeding, the reader might find the following quiz of interest:
| Table 1. Number of pharmaceutical products on the U.S. market as of January 1, 2002 5-7 | |||
| |||
NUMBER OF DRUGS ON THE U.S. MARKET AND THE IMPLICATIONS
As of January 1, 2002, there were over 3,200 prescription drugs, 600 herbal products, and 300 dietary supplements on the market in the United States (Table 1). This number does not include all available OTC medications. These numbers provide a measure of the complexity facing the prescriber, the patient, and the healthcare system when considering the issue of DDIs. It is this base to which new products are added.
A brief review of the findings of a study done in the Veterans Administration (VA) healthcare system will put these numbers in perspective.8,9 This study involved a survey of the extent and nature of prescription polypharmacy in approximately 5,000 patients (primarily outpa-tients) in one district of the VA healthcare system. Among the 5,000 patients, 400 different drugs were being prescribed. Although 400 is a substantial number, that is only approximately 10% of the number available to the U.S. prescriber (Table 1).
The study also found that the average VA patient was taking five different medications. In an earlier column,10 it was pointed out that 650 billion different potential combinations are possible if a prescriber starts with 600 drugs and uses up to 5 in combination. The VA study found that 75% of the VA population was on a unique combination of medications. In other words, 75% of those patients differed from each other (i.e., variable 3 in equation 1) based solely on the medications that they were taking. That is without considering interindividual differences due to age, genetics, or intercurrent disease.
The numbers in Table 1 reflect the potential for unique combinations in the general U.S. population as opposed to a closed healthcare system like the VA, which has a restricted formulary. As new drugs come to the US market, they add to this potential. If the entire formulary of 3,200 drugs on the U.S. market were available to a group of prescribers treating a patient and they used 5 drugs to treat that patient, the patient could be on one of 2.79 x 10 15 potential combinations.* Adding one new drug to the formulary would increase the number of possible combinations by 4.37 trillion. That is quite a bit to keep in mind.
DRUG DEVELOPMENT AND INTERACTION DATA
The drug development process typically provides only extremely limited data about how the new drug interacts with existing drugs. The number of formal DDI studies done during a drug’s development rarely exceeds 10. Moreover, those DDI studies are limited to the potential interaction between the new drug and just one of those other 10 drugs at a time. They do not address what the new drug does in the presence of four other drugs, which is the number of drugs the average VA patient is taking. In fact, there are virtually no formal DDI studies of what any drug does in combination with 4 other drugs. A few multiple drug DDI studies have been done principally in the areas of epilepsy and human immunosuppressive virus (HIV) because the usual standard of care for treating these conditions involves the use of combination drug therapy. But even for drugs used to treat these conditions, the studies are generally limited to the effects of three drugs in combination.
In addition to the data from formal DDI studies done during a drug’s development, experience is gained through concomitant drug treatment that is permitted during the efficacy trials of the new drug. However, that experience is limited by the protocol exclusion criteria governing the concomitant drugs a patient can be taking and still remain eligible for the study.
In antidepressant and antipsychotic clinical trials, the protocols generally contain a relatively short list of approved concomitant drugs, a list that includes far fewer drugs than the 400 found in the VA study and substantially fewer than the 3,200 drugs currently on the market in the United States. Also, in such studies, the patient cannot generally be on any other CNS active medication besides the study medication and the dose of any maintenance drug (e.g., an antihypertensive) the patient is taking must have been stable for at least 3 months prior to enrollment in the study. Patients also can have no unstable medical conditions (e.g., insulin dependent diabetes).
These exclusion criteria are put into efficacy trials of investigational antidepressants and antipsychotics to ensure that the benefits that are seen in the study are due to the study drug and not to a concomitant medication and to decrease the likelihood of a clinically significant adverse event occurring in the study due to an unstable medical condition. However, these criteria also limit the DDI experience with the new drug prior to its release on the U.S. market.
As a result of these exclusion criteria, the minority of patients in efficacy trials for investigational antidepressants and antipsychotics are on concomitant drug therapy. Most patients are only taking the investigational drug. Those who are taking a concomitant drug are generally only taking one other medication and often only intermittent therapy for a brief intercurrent condition such as sinusitis. Parenthetically, the experience with concomitant medication use is typically much greater with investigational anticonvulsants and HIV drugs than with investigational antidepressants and antipsychotics because the design of at least the early efficacy studies for investigational anticonvulsants and HIV drugs typically involves testing the efficacy of the investigational drug as add-on therapy to the existing medication regimen the patient is taking. This is done because these are chronic conditions and a sizable percentage of patients who do not adequately respond to existing therapy nevertheless have had a sufficient response so that it would not be ethical to "wash" the patient off of their existing regimen to test the efficacy of the investigational drug alone.
There are some negative consequences of such add-on studies, which are beyond the scope of this column but can be addressed in future columns. However, a number of the reasons that have led to their use in patients with epilepsy and HIV also apply to a sizable percentage of patients with major depression and schizophrenia. Both of those conditions can be chronic and a sizable percentage of patients with those conditions do not respond adequately to existing medications. Thus, including add-on studies in the portfolio of studies submitted to the U.S. Food and Drug Administration (FDA) to support the approval of investigational antidepressants and antipsychotics would be appropriate and would provide clinically useful information for the clinician in terms of the safety and efficacy of using the new drug in combination with existing medications in patients with chronic and partially treatment- refractory major depressive and psychotic disorders. The design of these studies would parallel those done with investigational anticonvulsants and HIV drugs. Such studies would provide at least a partial answer to one of the most frequent questions posed when a new antidepressant or antipsychotic comes to market: "Can I use this new drug in combination with drug X, Y, and/or Z (often all three)?"
| Figure 1. Number of drugs per year approved by the FDA for marketing in the United States 6,7 | |
|
DRUG APPROVALS
Figure 1 shows the number of drugs per year approved by the FDA for marketing in the United States from 1939 to 2000. As can readily be seen, the number has steadily increased over this period. On average, 3 drugs were approved per year in the 1940s, 10 in the 1950s, 11 in the 1960s, 17 in the 1970s, 28 in the 1980s, and 41 in the 1990s. In the last 5 years of the 20th century, almost 1 drug per week was approved for marketing in the United States. This means that, almost on a weekly basis, prescribers should be asking whether a new drug can be used safely and effectively in combination with other drugs the patient is taking, regardless of whether the practitioner is prescribing the new drug or one of the other drugs the patient is already taking.
There are a number of obstacles to the safe and effective incorporation of a new drug into clinical practice:
Silo Mentality
Prescribers may have a silo mentality so that they consider that their responsibility is limited to knowing the pharmacology only of the drug(s) they are prescribing. Although understandable, this is still a problem. Prescribers obviously need to be knowledgeable about the drugs they are prescribing because they bear responsibility for the consequences. However, Equation 1 shows that the consequences are determined in part by interindividual differences among patients, and as pointed out here and in many earlier columns, the other drugs a patient is taking are important determinants of interindividual differences. Remember that the goal of most drug therapy, with the exception of anti-infectives, is to change the biology of the patient taking the drug.11 That drug-induced change in the patient’s biology can alter his or her response to another drug, which is the essence of DDIs. Thus, prescribers cannot be concerned simply with the drug they are prescribing but must also consider what that drug will do in combination with the other drugs the patient is taking. It is analogous to taking into account whether the patient is a young child or an elderly adult (i.e., the age factor), whether there is seriously compromised cardiac, liver, or renal function (i.e., the disease factor), or whether there is a known genetic factor (i.e., variation) that can alter the pharmacodynamics or phar-macokinetics of the newly prescribed drug (e.g., cytochrome 2D6 deficiency).
Drug Education
After the completion of their formal training, most prescribers receive their education about new drugs primarily from representatives of pharmaceutical companies who call on prescribers and "detail" them on the new medication. This practice is not in and of itself a problem but it does mean that the source of information is limited in terms of both knowledge and perspective. This statement is made with an appreciation of the fact that pharmaceutical companies make tremendous efforts to have talented and well-trained representatives. Companies hire bright people and train them extensively. Often, although not invariably, these representatives have a biomedical background. A number are pharmacists and nurses. Nevertheless, these representatives are trained on the limited number of drugs that they detail and the competing products in that therapeutic arena. The typical pharmaceutical representative will have responsibility for perhaps six drugs in six therapeutic classes. Thus, they are susceptible to the same silo mentality as prescribers. This problem is compounded by the fact that, unless they have a clinical background, they may have a limited appreciation for the other factors (i.e., variable 3 in equation 1), which can shift the dose-response curve to their product. Being human, they also have a natural bias against accepting that one of those variables could cause a serious adverse response to their medication. After all, they should and typically do have faith in their company and the system (i.e., the drug development process and the FDA review) that led to the approval of their product. Their livelihood also depends on sales of their product. Finally, if physicians typically find it challenging to keep up with all the new drugs and potential new DDIs, why should pharmaceutical representatives be any different? This dis-cussion is not meant as a criticism but simply to point out the limitations of relying solely on this source of information.
Prescriber Targeting
Pharmaceutical companies, like any good businesses, naturally target their efforts to customers who are likely to prescribe their new product (e.g., practitioners in that therapeutic area). If a company produces an antibiotic, it is first going to focus its efforts on infectious disease specialists who have the potential to be high volume prescribers of the product and who may also have influence with formulary committees and with secondary target users such as primary care physicians. They are not likely to target psychiatrists, since the average psychiatrist does not usually prescribe antibiotics. However, that psychiatrist may be prescribing a drug that affects or is affected by the antibiotic to a clinically significant degree.4
It does not matter to the body who prescribed the drug nor why it was prescribed nor to what therapeutic class it belongs; all that matters are the drug’s pharmacodynamics (variable 1 in equation1) and pharmacokinetics (variable 2 in equation1), which in turn determine whether the drug will interact with other drugs the patient is taking. To gain a greater appreciation for this issue, the interested reader is referred to earlier columns that reviewed the complex interactions between multiple drugs, particularly the column detailing the complex interaction between the antibiotic, erythromycin, and codeine, metoprolol, and paroxetine.4 Due to the focused educational efforts of pharmaceutical companies, conscientious prescribers may not even hear about a new drug if they rely solely on this source of information rather than using sources such as the Medical Letter.
| Figure 2. Number of drugs withdrawn per year from the U.S. market 13,14 | |
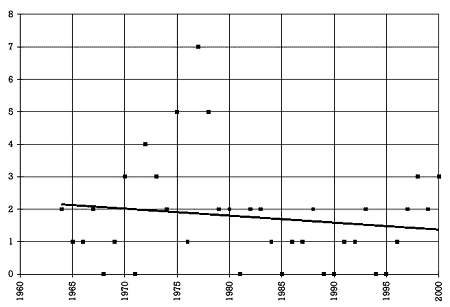 |
Potential Dangers: A Secondary Message
Pharmaceutical companies also naturally gear their efforts more to why the likely potential prescriber should use the new medication than to the potential dangers or limitations in the knowledge base concerning issues such as DDIs. Of course, companies do present the limitations of their products and the precautions that the prescriber should take—however, that message is secondary rather than primary. Obviously, the company would not have developed the drug nor would the FDA have approved it if they thought its dangers outweighed its benefits. Moreover, the company has made a substantial financial and to some extent emotional commitment to a drug in bringing it to the market. The financial commitment averages $500,000,000. This investment can only be recouped by selling the drug, which means the drug must stay on the market. The company has put its reputation on the line to some extent by bringing the drug to the market; it is also likely that one or more than one individual within the company was a champion for the drug (i.e., the emotional commitment). For these reasons, the company has a substantial belief about why prescribers should be using the drug rather than why under specific circumstances (e.g., the co-prescription of another drug) they should not. The prescriber may in fact learn more about the limitations of a new product from its competitors rather than the manufacturer. However, that information is not without obvious biases and the question then arises whether the competitor is making much ado about nothing. The reader is referred to earlier columns on the issue of CYP enzyme inhibition and SSRIs as an example of this phenomenon.1,12
DRUG WITHDRAWALS
The fact that fluoxetine and paroxetine are on the market raises the question of how foolproof the current drug development and registration process is. Figure 2 shows the number of drugs withdrawn from the U.S. market over the last 37 years. During this period of time, an average of 1.8 drugs were withdrawn every year from the U.S. market. This is almost equal to the average number of drugs approved per year for the U.S. market in the 1940s. Some prescribers may find that fact surprising. They may be even more surprised to learn that in only 8 out of the last 37 years was no drug removed from the market. Thus, historically there has been almost an 80% chance that at least one drug would be withdrawn in a given year.
Does that mean the drug development and registration process is seriously flawed? No. Instead, it illustrates the wisdom behind the saying "Nature sides with the hidden flaw."
Like most facets of drug development and marketing, the issue of drug withdrawals is not without its hype. To test that claim, interested readers can go to the Internet and enter "drug withdraw" as a search term. They will be rewarded with multiple hits for websites from a diverse range of sources, including the FDA, pharmaceutical companies, attorneys, Wall Street brokerage houses, and various consumer groups. Although each of these sites has its own agenda, the sites illustrate the varied and substantial interest in this issue. Some of these sites suggest that the withdrawal of any drug is evidence of the inadequacy of the approval process; however, these sites are likely to come from either uninformed or self-serving sources.
Pharmaceutical companies and the FDA are faced with challenges similar to those facing the prescriber. The basic problem is a lack of relevant knowledge at the time the drug was being developed and approved. Again, fluoxetine and paroxetine and their inhibition of one or more CYP enzymes are examples of this fact. At the time those drugs were developed, there was no reason or way to screen for unintended effects of CYP enzymes. Now it is possible and such screening has become a standard part of the drug development process. As noted above, these drugs would probably not make it out of preclinical drug development today.
| Table 2. Drugs approved and withdrawn from the U.S. market over the last 37 years6,7,13,14 | |||||||||
| |||||||||
The fact that the system is not flawed and is in fact improving on a yearly basis is shown in Table 2. While the absolute number of drugs per year withdrawn from the U.S. market has not declined over the last 37 years, the number of drugs withdrawn relative to the number of drugs approved has. This ratio was 12.5% in the 1960s, 19% in the 1970s, and only 4% in the 1980s and 1990s. Of course, a caveat is that the higher rate in the early years was also a function of cleaning up drugs that had been approved in an era when both the drug development and approval process were less rigorous. Nevertheless, the current process does in general work remarkably well.
A review of the drugs withdrawn from the market shows that the system fails in certain specific areas, some of which are built into the drug development process itself, while others are the result of a lack of knowledge.13,14 For example, a lack of knowledge is reflected in the fact that most of the drugs withdrawn during the last several years were taken off the market because of CYP enzyme-mediated DDIs that had not been appreciated prior to the experience with terfenadine and Torsades de Pointes. While most of the drugs that were withdrawn were the victims of the interaction, mibefradil (Posicor), which is a substantial CYP inhibitor, was withdrawn because it was the perpetrator of the interaction, having been found to cause potentially dangerous elevations of at least 26 other drugs frequently co-prescribed with it.
The cases of nomifensine, dexfenfluramine, encainide, and temafloxacin illustrate limitations that are to some extent built into the current drug development process.
Nomifensine was a novel antidepressant marketed in the United States in the late 1980s. While a useful antidepressant for some patients, it was withdrawn from the U.S. market after less than a year because it caused Coombs positive hemolytic anemia. The risk of this adverse effect increased as a function of recurrent courses of treatment. One problem with the current approach to drug development, at least for antidepressants and antipsychotics, is that a patient can be entered into a trial with the medication only once. There is a good reason for this stipulation but it effectively prevents the detection of problems, such as the hemolytic anemia produced by nomifensine, that become apparent only with a second or third exposure to the drug after having been off it for a period of time.
Dexfenfluramine was a weight-reducing medication that was claimed to be linked to the development of cardiac valvular insufficiency after chronic exposure to the drug when taken in combination with phentermine. While some studies in its new drug application involved treatment for up to 1 year, the duration of treatment and the number of patients exposed were insufficient to detect this adverse effect.
Encainide was an anti-arrhythmic that could successfully suppress ventricular arrhythmias and thus increase survival in patients who had such arrhythmias following an acute myocardial infarction. However, it was withdrawn when it was found to increase the mortality rate if the patient had a second infarction while on the drug. The studies done to support registration were too short to detect this problem.
Temafloxacin, a fluoroquinolone antibiotic, was withdrawn after it was found to cause an increased risk of multiple and varied serious adverse effects in elderly patients. Of note, only a few hundred "young" elderly (i.e., individuals 70 years or younger) are typically included in the clinical trials of most new approved drugs and the inclusion/exclusion criteria require that these be healthy "young" elderly.
Other problems that have led to the withdrawal of drugs over the past 37 years are essentially not detectable in any practical way with current knowledge or techniques. For example, years after diethylstilbestrol, a synthetic hormone used to prevent premature delivery, was approved, it was found to cause an increased incidence of adenocarcinoma in the female offspring of patients who received the drug.
PROBLEMS POSED BY DRUG WITHDRAWALS AND POTENTIAL SOLUTIONS
An earlier column made the point that the goal of the drug development process is to reduce uncertainty about a drug’s effect. It does not make the drug safer but simply defines its risk relative to its benefits. That information is first used to determine whether the drug should be approved. It is also used to inform the prescriber about how to use the drug safely (i.e., one of the goals of the package insert). While the drug develop-ment and approval system is remarkably good and getting even better, drug withdrawals are examples of its fallibility.
There are of course practical limits to what can be done in the drug development process. One obvious limitation is the extent of our knowledge at the time a drug is developed. Another is practicality. The drug development process cannot last so long that the patent life expires prior to marketing. If that occurs, then it becomes commercially impractical to develop the drug. In fact, this situation led to a change in the patent laws so that a sponsor now has 5 years of exclusivity once a drug has been approved even if the drug’s patent life has expired. If that change had not been in place, neither citalopram nor mirtazapine would have made it to the market, since the patent life had expired prior to the drug’s approval in both of these cases.
The saying "time is money" is certainly true for drug development. The more time used in the drug development process, the less time there is to sell the drug while under patent to recoup expenses and hopefully realize a profit. Reducing development time is a major goal for pharmaceutical companies. One hope for the human genome project is that it will lead to ways of reducing drug development time as well as increasing the likelihood of success by identifying both novel targets to hit (variable 1 in equation 1) and targets to avoid.1 In addition to consuming valuable patent life, a longer development time usually means that more studies have been done and hence more money has gone into the development—which raises the issue of how many and what types of patients need to be studied and for how long. These issues are relevant to what occurred with drugs such as dexfenfluramine, encainide, temafloxacin, and diethylstilbestrol. Obviously, it is impractical to require a drug to be in development for 20 years or longer to see if it has an effect on the second generation. Instead, animal studies are used to test for such effects in the preclinical phase of the drug development. The success of such studies depends on how similar the drug’s effect is in the animal species studied and man.
With all of the above caveats, it is in everyone’s best interest (perhaps with the exception of the plaintiff’s attorney) to avoid the development and approval of drugs that will subsequently be pulled from the market because of safety reasons. From a company’s standpoint, the downsides are obvious and include loss of revenue, loss of confidence by prescribers and the public, and the possibility of lawsuits. In terms of lost revenue, the New York Times estimated that seven of the drugs most recently withdrawn from the U.S. market were generating $5 billion in sales annually and that their sales were still growing.15 These sale figures suggest that these drugs were fulfilling some need, which in turn explains why they were approved in the first place. When the drug has been pulled from the market, it can no longer fulfill that need. This raises another less obvious problem with drug withdrawals: How significant is the unmet need? Are there other agents that can do just as well? Or did the withdrawn drug produce a truly unique benefit in some segment of the population? In this instance, one is sometimes weighing the relative risk to a few versus the relative benefit to many. This issue is particularly difficult when the adverse event is quite rare and there is no obvious pattern to suggest how to avoid it.
Alostetron (Lotronex) and nefazodone are two examples of drugs that were recently discovered to cause rare adverse effects.
Alostetron, a potent and selective antagonist of the serotonin 5-HT3 receptor, was recently pulled from the U.S. market after 70 of the 300,000 (0.02%) patients taking this drug developed ischemic colitis, a risk of 2 out of 10,000 patients. While not withdrawn from the market, nefazodone has recently received a black box warning.16 The addition of a black box warning is generally the most restrictive regulatory change that can be made short of withdrawing a drug from the market. The severity of a black box warning is reflected in the fact that Abbott Laboratories decided against marketing sertindole because it was approved with a black box warning. The black box warning for nefazodone was occasioned by the occurrence of one case of liver failure resulting in death or the need for liver transplantation per 250,000 to 300,000 patient-years of nefazodone treatment. The recent experience with alostetron and nefazodone illustrate several important points.
First, a drug development program would have to involve tens of thousands of patients to detect such rare adverse events with any confidence. In contrast, most antidepressant drug development programs involve the exposure of a few thousand patients. For example, the nefazodone program involved a total of 3,496 patients.17 In addition to the fact that the patients enrolled in such trials are carefully selected to be medically healthy and to be on few, if any, other medications, they generally receive only short-term (a few weeks) treatment. In the case of nefazodone, 250 out of the 3,496 patients were treated for at least 1 year. In contrast, most patients who received these two drugs once they were marketed received them chronically.
These examples also raise two questions:
The answers to these questions lie in the third variable in equation 1, interindividual variance, which accounts for differences in drug response including rare adverse effects. In the case of these two drugs, there were no obvious differences in age or intercurrent disease between those who developed these adverse effects and those who did not. There was also no obvious drug combination that predicted these adverse effects, although that may simply be due to the small number of cases.
Another potential explanation for these adverse effects would be genetic susceptibility or some combination of the various factors described above. DNA samples could be obtained from as many of the 70 patients who developed ischemic colitis on alostetron as could be located and were willing to give consent. These samples could then be screened for a genetic polymorphism that distinguished them from the 9,998 who did not develop ischemic colitis despite taking comparable amounts of alostetron for a comparable period of time. If a genetic risk factor could be identified, then conceivably the drug could be returned to the market with a label that instructed prescribers to first test patients for this genetic risk factor before treating them with alostetron. Such an approach would provide for the safety of those with the risk factor, while allowing the other 9,998 individuals who are not at risk to receive a potentially therapeutic trial of the medication, and would allow the manufacturer to recoup and potentially make a profit on its investment in the development of the drug. There are in fact companies such as Genaissance Pharmaceuticals who have the capability to do just such testing. Thus, the human genome project has the potential to rehabilitate problem drugs and return them to the market in addition to providing new targets for drug discovery and new means of more rapidly and efficiently conducting high-yield drug development programs. The above scenario is not science fiction—it has already been successfully applied to understanding why some patients are genetically predisposed to reactions to sulfa drugs.
The answers to the short quiz presented earlier in the column are given below. It is hoped that this column will help clinicians better appreciate the complexity of prescribing medications, particularly for the patient who is already on multiple medications, and the need to be on the lookout for potential adverse effects.
ANSWERS TO THE QUIZ
* 3200!/[5!(3200-5)!] = 2.79 x 1015 See "Do you feel lucky?" 10 for a discussion of the formulas used to calculate these numbers.