|
|
Clinical Pharmacology of SSRI's
Tables and Figures |
|
|
|
|
|
As discussed in Section 8, the in vitro inhibition
constant (Ki) is not the sole determinant of whether a drug
will produce a clinically meaningful or even detectable in
vivo effect on a cytochrome P450 (CYP) enzyme under clinically
relevant dosing conditions. Instead, it is only one factor
in the equation:
Equation 1:
Magnitude
of Effect |
= |
Affinity for Site of Action x Drug
Concentration of Effect at Site of Action x Underlying
Biology of Patient |
For this reason, the Ki has relatively limited
value without knowing or at least having a reasonable estimate
of the concentration of the potential inhibitor at the site
of action (eg, the CYP enzyme). Parenthetically, the location
of the relevant CYP enzyme may be extrahepatic in some instances,
such as in the gut wall in the case of the inhibition of the
first pass metabolism of terfenadine.
Although some reviews have attempted to rank or list drugs
as enzyme inhibitors based on primarily in vitro data
or isolated case reports of possible interactions,2
this approach does not convey to the clinician whether an
interaction is likely to occur in clinical practice or to
what extent. Answers to these questions require knowing the
concentration of the drug that will occur under clinically
relevant dosing conditions as well as the Ki.
This appendix is for the reader who would like more background
on this issue from a mathematical perspective, but is not
intended to be a rigorous mathematical treatise on the subject.
This discussion is relevant to any drug or class of drugs
capable of inhibiting a CYP enzyme; however, it will use the
SSRIs as the example since they are the subject of this book.
The reader who would like further discussion of these matters
is referred to the work of Segel3 and von Moltke
and colleagues.4
For this discussion, we must recall some basic principles
of biochemistry, specifically enzymology. The relationship
between the inhibition of an enzyme caused by a competitive
inhibitor and the concentration of the inhibitor is expressed
by the equation:
Equation 2:
i = [I] / ( [I] + Ki (1 + [S] / Km)
)
where “i” is the fractional inhibition of the enzyme,
[I] is the concentration of the inhibitor, Ki is
the inhibition constant of the inhibitor for the enzyme, [S]
is the concentration of the substrate normally biotransformed
by the enzyme, and Km is the affinity constant
of that substrate for that enzyme.
If [S] is less than Km as is the case in vivo
for drugs with linear pharmacokinetics,then the fractional
inhibition is independent of the substrate concentration and
the equation reduces to:
Equation 3:
i = [I] / ( [I] + Ki )
Hence, to the first approximation, the fractional inhibition
of the enzyme is determined by the concentration of the inhibitor
directly and by the Ki of the inhibitor reciprocally.
The smaller Ki is to [I], the more “i”
approaches unity:
Equation 4:
i = [I] / [I] (when Ki <<<< [I])
Equation 4 illustrates why both Ki and [I] are
important when trying to determine whether a clinically meaningful
degree of enzyme inhibition is likely to occur under clinically
relevant dosing conditions. Parenthetically, the relative
Ki’s in some studies (eg, Crewe et al, 1991)
can be outliers relative to the ones derived from other in
vitro studies (Table 8.7). If the concentration of the
substrate used in the study not being much less than its Km
and not reflective of what would be expected in vivo.
Since all of the SSRIs except possibly fluvoxamine have
“active” metabolites in terms of the inhibition
of specific CYP enzymes such as CYP 2D6, one must have equation
3 for the parent drug and all of its relevant metabolites
when doing projections from in vitro work to in
vivo reality. Parenthetically, formal in vivo pharmacokinetic
studies measure the summed effect of the parent drug and all
of the relevant metabolites at the relative concentrations
that they occur under clinically relevant dosing conditions,
assuming that the drug is given as it would normally be given
and given for a sufficient period of time to reach steady-state
conditions.
This last condition has not been true for many of the fluoxetine
studies reviewed in Section 8. Instead, loading dose strategies
(eg, 60 mg/day × 8 days) have commonly been used due
to the long period of time that the study will have to go
to reach steady-state conditions of fluoxetine and norfluoxetine.
The results of such loading dose strategies may underestimate
the actual effect that will occur under more clinically relevant
dosing conditions for several reasons.
First, norfluoxetine is more potent than fluoxetine in terms
of the inhibition of some CYP enzymes such as CYP 3A3/4. Secondly,
the conversion of fluoxetine to norfluoxetine takes time.
Third, this conversion is inhibited by high concentrations
of fluoxetine and/or norfluoxetine. Hence, such loading dose
approaches may underestimate the ratio of norfluoxetine to
fluoxetine that will be expected under truly steady-state
conditions and thus underestimate the concentration of the
more portent CYP 3A3/4 inhibitor, norfluoxetine. For these
reasons, the results from the loading studies with fluoxetine
must be interpreted cautiously and do not reflect the effect
that will occur under steady-state conditions at at 20 mg/day,
much less 60 mg/day.
The next step in trying to relate in vitro results
to in vivo reality is to determine what is the relevant
concentration at the CYP enzyme. The concentration of the
drug in plasma is the most readily measured concentration;
however, the enzyme is not in the plasma, but rather in some
tissue compartments. The hepatic tissue compartment is the
one which is generally most relevant to predicting whether
a pharmacokinetic drug-drug interaction is likely to occur
due to the inhibition of a CYP enzyme. For this reason, we
need to either measure or estimate the hepatic concentration
of the drug which occurs in this compartment under clinically
relevant dosing conditions.
Typically, the hepatic concentration is estimated rather
than directly measured. It is estimated based on measuring
the plasma drug concentration of the drug and its plasma:liver
partition coefficient (Table 10.1). The plasma:liver partition
coefficient is empirically determined in animals and then
can be confirmed in man using autopsy or surgical material.
Table 10.1 illustrates how such information can be used
to estimate the relative inhibition of a specific CYP enzyme
using such information. Table 10.1 compares the estimated
inhibition of CYP 2D6 by 4 different SSRIs. First, the average
Ki for each SSRI was calculated using the data
from the 5 in vitro studies reported in Table 8.7.
Since all 5 of these studies measured the Ki for fluoxetine
but not for all the other SSRIs, the average Ki
for fluoxetine was determined and used to normalize the Ki
values for all the other SSRIs in terms of their relative
value compared to fluoxetine. The plasma levels of the different
SSRIs which would be expected under comparable antidepressant
treatment conditions was then determined. The hepatic concentration
of each SSRI which would be expected under such conditions
was then estimated based on each drug’s plasma:liver
partition coefficient times their expected plasma drug concentration
at steady-state under comparable antidepressant treatment
conditions. These values
and equation 2 above correctly predict the substantial differences
in the degree of CYP2D6 inhibition which has been measured
in the 11 formal in vivo pharmacokinetic studies reviewed
in Section 8 on the relative effects of these SSRIs on CYP
2D6 function. As can be readily seen, a comparison using the
Ki alone will lead one to erroneously conclude
that there may be minimal differences between these different
SSRIs with regard to the inhibition of this enzyme under clinically
relevant conditions.
The next issue is how the fractional inhibition of the enzyme
relates to the change in the concentration of a concomitantly
administered drug which is dependent on the functional integrity
of this enzyme for its clearance. In clinical practice, the
term “concentration” usually refers to either a
12-hour post-dose concentration or a trough concentration
(ie, the concentration immediately before the next dose of
drug is given). In both instances, these concentrations are
typically measured after steady-state has been attained or
assumed to have been attained. A more rigorous measurement
is to measure the area under the plasma concentration-time
curve (AUC) under steady-state conditions. The latter approach
is typically used in formal pharmacokinetic studies. Nonetheless,
both approaches have been used to assess the change in the
drug concentration as a function of the decrease in its clearance
produced by the inhibition of the principal CYP enzyme responsible
for the biotransformation necessary for the drug’s elimination.
The increase in the AUC with the inhibitor present (AUCi)
relative to the AUC without the inhibitor present is related
to the fractional inhibition of the enzyme (i) as follows:
Equation 5:
AUCi/AUCo = I/ ( 1-I )
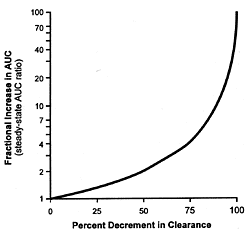
As can be readily appreciated, equation 5 describes a complex
hyperbolic relationship (Figure 10.1) between the fractional
inhibition of the enzyme and the change in the plasma concentration
of the drug whose metabolism has been inhibited. This curve
approximates linearity over narrow portions of the curve.
As the inhibitions increase, the increase in the plasma concentration
of the affected drug increases disproportionately. This fact
further explains the differences in clearance of drugs such
as desipramine which are observed when fluoxetine and paroxetine
are coprescribed at their usually effective minimum antidepressant
doses versus citalopram, fluvoxamine, and sertraline (Table
10.1). Fluoxetine and paroxetine produce substantially more
than 50% inhibition of the enzyme in contrast to the other
3 SSRIs and thus are on the rapidly ascending portion of this
hyperbolic curve.
As stated at the beginning, this discussion has used the
effect of the SSRIs on CYP 2D6 to illustrate the enzymological
and pharmacokinetic principles relevant to understanding in
greater depth the differential effects of drug-induced inhibition
of CYP enzyme-mediated clearance of concomitantly prescribed
drugs. These principles are relevant to the effects of the
SSRIs on other CYP enzymes and to the
effects of other drugs on the various CYP enzymes.
| TABLE
10.1 Variables Which Determine the Magnitude of the
Effect In Vivo: CYP 2D6 as an Example |
| |
Fluvoxamine |
Fluoxetine |
Paroxetine |
Sertraline |
| Relative Ki |
9.4 |
1.0 |
0.8 |
6.4 |
| Estimated Ki (µM) |
8.31 |
0.88 |
0.71 |
5.66 |
| Plasma concentration (nM) |
164.0 |
512.0 |
135.0 |
66.0 |
| Liver:water partition ratio |
26.6 |
12.1 |
26.2 |
12.2 |
| Liver concentration (µM) |
4.4 |
6.2 |
3.5 |
0.8 |
Change in desipramine*
PK studies (AUC) |
14% |
380% to 640% |
327% to 421% |
0% to 37% |
| * Above baseline. |
| AUC = Area under
curve. |
| References: 120 |
| FIGURE 10.1 |
 |
Using the approach
explained above, in vitro studies can be used to screen
already marketed drugs or drugs in the development for their
effects on CYP enzymes and to predict using a knowledge of
their Ki, the plasma drug concentration expected
under clinically relevant dosing conditions and the plasma:liver
partition coefficient whether a clinically meaningful change
in the clearance of specific drugs can be expected if they
were concomitantly administered with the potential inhibitor.
Based on such in vitro modeling, appropriate in
vivo studies can be done on a selective basis to confirm
such predictions when such an interaction would be predicted
to occur in a significant number of patients (ie, the drugs
are likely to be frequently coprescribed together) and the
consequences are predicted to be clinically important. The
advantage of this sequential in vitro and then in
vivo approach is the cost and time efficiency of doing
in vitro rather than in vivo studies to screen
the drugs against a full battery of CYP enzymes.
Given this discussion, readers can anticipate
that they will be hearing more and more about the effects
of a wide variety of drugs on CYP enzymes. This information
will aid the physicians in anticipating pharmacokinetic interactions
and will allow them to make appropriate treatment decisions
(eg, drug selection, dose adjustments) to avoid adverse consequences
of such interactions. The SSRIs and their differential effects
on CYP enzymes have been a significant impetus and vehicle
for educating physicians about this important advance in our
knowledge.
| TABLE 10.2 SSRIs:
Brand Names by Country |
| Country |
Citalopram |
Fluoxetine |
Fluvoxamine |
Paroxetine |
Sertraline |
| Argentina |
-- |
Animex-On
Equilibrane
Foxetin
Neupax
Saurat |
-- |
Aropax |
Zoloft |
| Australia |
-- |
Prozac 20 |
-- |
Aropax |
Zoloft |
| Austria |
Seropram |
Fluctine |
Floxyfral |
Seroxat |
Tresleen |
| Belgium |
Cipramil |
Prozac |
Floxyfral |
Aropax
Seroxat |
Zoloft |
| Canada |
-- |
Prozac |
Luvox |
Paxil |
Zoloft |
| Denmark |
Cipramil |
Fontex
Fonzac |
Fevarin |
Seroxat |
Zoloft |
| Finland |
Cipramil |
Fontex
Seronil |
Fevarin |
Seroxat |
Zoloft |
| France |
Cipramil |
Prozac |
Floxyfral |
Deroxat |
Zoloft |
| Germany |
Saroten |
Fluctin |
Fevarin |
Seroxat
Tagonis |
Zoloft |
| Greece |
Seropram |
Flonital
Fluxadir
Ladose
Orthon |
Dumyrox |
Seroxat |
-- |
| Italy |
-- |
Fluxeren
Prozac |
Dumirox
Fevarin
Maveral |
Sereupin
Seroxat |
Serad
Tatig
Zoloft |
| Mexico |
-- |
Fluoxac
Prozac |
-- |
Aropax
Paxil |
Altruline |
| Netherlands |
-- |
Prozac |
Fevarin |
Seroxat |
Zoloft |
| Norway |
Cipramil |
Fontex |
Fevarin |
Seroxat |
Zoloft |
| Portugal |
-- |
Digassim
Nodepe
Prozac
Psipax
Tuneluz |
Dumyrox |
-- |
-- |
| South Africa |
Cipramil |
Prozac |
Luvox |
Aropax 20 |
Zoloft |
| Spain |
-- |
Adofen
Prozac
Reneuron |
Dumirox |
Frosinor
Motivan
Seroxat |
Aramis
Besitran |
| Sweden |
Cipramil |
-- |
Fevarin |
Seroxat |
Zoloft |
| Switzerland |
Seropram |
Fluctine |
Floxyfral |
Deroxat |
Zoloft
Gladem |
| Turkey |
-- |
Depreks
Prozac |
Faverin |
-- |
Lustral |
| United Kingdom |
-- |
Prozac |
Faverin |
-- |
Lustral |
| United States |
-- |
Prozac |
Luvox |
Paxil |
Zoloft |
TABLES
Table 2.1
Major Classes of Antidepressants Defined by Principal Mechanisms
of Action 16
Table 2.2 Criteria
for New Drug Development 19
Table 2.3 The Evolution
of Psychopharmacology 20
Table 2.4 TCA (Amitriptyline)
Polypharmacy in a Single Pill 25
Table 3.1 Effect
of Antidepressants on Serotonin Uptake In Vitro 37
Table 3.2 Effect
of Antidepressants on Norepinephrine Uptake In Vitro 38
Table 3.3 Effect
of Antidepressants on Dopamine Uptake In Vitro 39
Table 3.4 Relative
Potency of the Enantiomers of Citalopram, Fluoxetine and Their
Metabolites for Inhibiting the Uptake Pumps for Different
Biogenic Amine Neurotransmitters 44
Table 3.5 In Vitro
Selectivity Ratio for Different SSRIs and Selected TCAs 46
Table 3.6 Pharmacologic
Properties of Antidepressants and Possible Clinical Consequences
48
Table 3.7 Relationship
Between Dose, Plasma Level, Potency and Serotonin (5-HT) Uptake
50
Table 3.8 Effect
of Uptake Inhibitors and Their Metabolites In Vitro 58
Table 3.9 Effect
of Metabolism on the Central MOA and Half-lives of Some SSRIs
60
Table 4.1 STEPS:
Factors to Be Considered When Selecting a Medication for a
Patient 64
Table 4.2 Safety
and Tolerability of TCAs Versus SSRIs 65
Table 4.3 Placebo-adjusted
Incidence Rate (%) of Frequent Adverse Effects on Imipramine
68
Table 4.4 Response
Rates in Patients With Major Depressive Disorder by Meta-analysis
71
Table 5.1 Common
Features of SSRIs With Regard to the Treatment of Major Depression
75
Table 5.2 Comparison
of the Placebo-adjusted Incidence Rate (%) of Frequent Adverse
Effects for SSRIs 82
Table 5.3 Adverse
Events for Each SSRI That Occur ³ 1% More Often Than
With Other SSRIs 86
Table 5.4 Placebo-adjusted
Incidence (%) of Various Forms of Sexual Dysfunction on Four
SSRIs 88
Table 5.5 SSRI Versus
Placebo: Response Rate and Relapse Rate 92
Table 6.1 Concentration-dependent
Effects of SSRIs 108
Table 6.2 Pharmacokinetic
Parameters Relevant to the Use of SSRIs 109
Table 6.3 CYP Enzyme
Responsible for Biotransformation of SSRIs 111
Table 6.4 Change
in Half-life (t1_2) as a Function of Multiple Dose Administration
118
Table 6.5 Effects
of Dose and Age on the Plasma Levels of SSRIs 120
Table 6.6 Effect
of Liver Disease on SSRI Metabolism and Pharmacokinetics 127
Table 6.7 Effect
of Renal Impairment on Pharmacokinetics of SSRIs (Single Dose)
127
Table 7.1 Unintended
Targets of Some SSRIs: CYP Enzymes 131
Table 7.2 History
of Our Knowledge of Biotransforming Enzymes 133
Table 7.3 Human
CYP Enzymes as Classified by Family, Subfamily and Gene 134
Table 7.4 Two General
Classes of CYP Enzymes 134
Table 7.5 What Are
the Functions of CYP Enzymes? 136
Table 7.6 Types
of Drug Interactions 145
Table 7.7 Pharmacokinetic
Interactions: How Do They Present Clinically? 146
Table 7.8 Examples
of Metabolically Mediated Pharmacokinetic Drug-drug Interactions
and Their Clinical Presentations 152
Table 7.9 Drugs
Metabolized by CYP Enzymes 158
Table 7.10 Genetically
Determined CYP Enzyme Deficiency 160
Table 8.1 Prevalence
of Major Depression in Specific Medically Ill Populations
162
Table 8.2 Concomitant
Use of Antidepressant With Other Medications in Different
Patient Populations 162
Table 8.3 Could
Inhibition of CYP Enzymes Have Other Consequences? 164
Table 8.4 Some SSRIs
Lack Selectivity With Regard to Effects on Serotonin Uptake
Versus CYP 2D6 165
Table 8.5 Common
Myths About CYP Enzymes and SSRIs 166
Table 8.6 Drug-induced
Inhibition of CYP Enzymes 167
Table 8.7 The Relative
Potency of Five Different SSRIs and Their Metabolites for
Inhibiting the Functional Integrity of Three CYP Enzymes 1A2,
2D6 and 3A3/4 Based on In Vitro Studies Using Human Hepatic
Microsomes 170
Table 8.8 Effects
of Specific SSRIs on Specific CYP Enzymes at Their Usually
Effective Antidepressant Dose 176
Table 8.9 In Vivo
Studies of Effects of Different SSRIs on CYP 2D6 Function
179
Table 8.10 Relative
Potency of the Enantiomers of Fluoxetine and Norfluoxetine
for Inhibiting the CYP Enzyme 2D6 180
Table 8.11 Comparison
of the In Vivo Effects of Different SSRIs on Specific CYP
Enzyme Substrates 193
Table 10.1 Variables
Which Determine the Magnitude of the Effect In Vivo: CYP 2D6
as an Example 230
Table 10.2 SSRIs:
Brand Names by Country 234
FIGURES
Figure 2.1 Structural
Formulas of Several SSRIs 17
Figure 2.2 Schematic
Illustration of Relationship Between Drug Site of Action and
Effect 19
Figure 2.3 Standard
and New Generation Antidepressants Mechanisms of Action 22
Figure 2.4 In Vitro
Potency of Amitriptyline as a Representative Tricyclic Antidepressant
for Different Sites of Action and Related Mechanisms of Action
27
Figure 3.1 Generic
Curve of a Drug's Concentration-dependent Effect on Specific
SOA 35
Figure 3.2 Selectivity
Ratios for a Series of Uptake Inhibitors Measured In Vitro
41
Figure 3.3 In Vitro
Profile of Antidepressants 53
Figure 4.1 Comparative
Incidence of Side Effects Between Amitriptyline and Sertraline
70
Figure 5.1 Discontinuation
Rate Due to Adverse Events as a Function of Dose for Three
SSRIs 84
Figure 5.2 Antidepressant
Efficacy as a Function of Dose for Three SSRIs 85
Figure 5.3 Relationship
Between Daily Dose of Sertraline, Mean Plasma Levels of Sertraline,
and Mean Reduction in Serotonin Uptake by Platelets After
14 Days of Drug Administration at One of Four Fixed Doses
97
Figure 5.4 Estimated
Minimum Effective Drug Concentration: Average Plasma Drug
Concentration Achieved in the Group Treated With the Minimum,
Effective Dose 103
Figure 6.1 Time
to Steady-state and Time to 95% Washout 123
Figure 7.1 Drug
Metabolism by CYP Enzymes 136
Figure 7.2 CYP Enzyme
Reaction Cycle 138
Figure 7.3 Diverse
Monooxygenase Activities of CYP Enzymes 139
Figure 7.4 Phases
of Xenobiotic Metabolism 140
Figure 7.5 Relationship
Between Dosing Rate, Clearance, Steady-state Drug Concentration,
and Clinical Response 141
Figure 7.6 Relationship
of Pharmacodynamics, Pharmacokinetics and Biological Variance
in Determining Overall Result of Drug Treatment 142
Figure 7.7 Dose-response
Curves With Risperidone 147
Figure 7.8 Dose-response
Curves for Seizure Risk With Clozapine and Bupropion 149
Figure 7.9 Multiple
Concentration: Response Curves of Tertiary TCAs 154
Figure 7.10 How
Knowledge of Drug-metabolizing Enzymes Will Simplify Understanding
of Pharmacokinetic Interactions 156
Figure 8.1 Design
of Pharmacokinetic Interaction Studies Using SSRIs as an Example
173
Figure 8.2 Differential
In Vivo Effects of Five Different SSRIs on CYP 2D6 Function
185
Figure 8.3 Trough
Concentrations of Desipramine in Plasma Correlated With Concentrations
of Sertraline Plus Desmethylsertraline in the Sertraline Treatment
Group and Fluoxetine Plus Norfluoxetine in the Fluoxetine
Treatment Group 188
Figure 8.4 Trough
Concentrations of Desipramine in Plasma Correlated With Concentrations
of Sertraline Plus Desmethylsertraline in the Sertraline Treatment
Group and Paroxetine Plus Norfluoxetine in the Paroxetine
Treatment Group 189
Figure 8.5 Relative
In Vivo Effects of CYP 3A3/4 Inhibitors on Triazolobenzodiazepines
195
Figure 10.1 Fractional
Increase in AUC/ Percent Decrement in Clearance 232
|



