|
|
| The Human Genome Project and Modern Drug Development in Psychiatry |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, September 2000, 272-276
|
This column is dedicated to Samuel B. Guze, MD, who died suddenly last month. Dr. Guze was perhaps the most influential psychiatrist in the last half of the 20th century. He, together with his colleagues at Washington University, were the driving forces in returning American psychiatry to its roots as a discipline based on the principles of science and medicine. He championed the use of operational criteria, epidemiological studies, and basic neuroscience research as ways of increasing our understanding of and ability to treat psychiatric illness. Dr. Guze's influence on the field is felt by all mental health workers every time they use our current Diagnostic and Statistical Manual of Mental Disorders, which is based on the principles he advocated. As a small tribute to this remarkable person, this column is the first written after the completion of the first full DNA sequencing of a human being (see Dr. Oldham's editorial, "The Genome Promise for Psychiatry," in the July issue of this journal). That development has enormous implications for our ability to understand and treat psychiatric illnesses. However, we would not be in a position to capitalize on this advance were it not for the foundation laid by Dr. Guze and his colleagues at Washington University.
In this column, I begin a series in which I will cover topics such as
- Microarrays ("gene chips")
- Orphan receptors
- High-through-put screening
- Model organisms
- Functional genomics
- Structure-activity relationships
- Molecular targeting
- Stages of drug development
- Reverse pharmacology
- Bridging studies
The knowledge derived from these approaches will permit us to enter the next stage of rational development of psychiatric medications and other somatic therapies. From this perspective, this column will extend our previous discussion contrasting newer versus older antidepressants to explain how even the most successful of the newest medications are simply another step on a rapidly accelerating evolutionary curve from chance to rational drug discovery.
The Evolution of Drug Development
In the column, "Marooned: Only One Choice," I pointed out that antidepressant drug development in psychiatry began with the chance observation that tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) had antidepressant activities. Both of these classes of drugs were initially developed unsuccessfully for other indications. Such chance discovery is not unique to psychiatry, but instead has been the rule for virtually all of the first drugs in every current major therapeutic class. Think, for example, of how Fleming and his moldy bread transformed the treatment of infectious disease.
In the series of columns I have just completed, we reviewed how the TCAs served as the blueprint for the development of the newer antidepressants. The selective serotonin reuptake inhibitors (SSRIs) have served in these columns as an example of a class of antidepressants developed rationally using 1970s technology. Each of the members of this class is truly a variation on a theme in terms of much but not all of its pharmacology. These drugs were in fact rationally developed to produce only one of the myriad effects of TCAs-the effect on serotonin uptake inhibition.
While a clear success story, the SSRIs are a product of what we knew and were able to do using 1970s technology, for the simple reason that all five SSRIs were synthesized and selected for development during the 1970s. This includes the most recently marketed SSRI, citalopram, which in 1972 was the second SSRI to be synthesized. Ironically, citalopram might have been the first rather than the last SSRI marketed in the United States if not for a cytochrome P450 enzyme in Beagle dogs. However, that story will need to wait for another column.
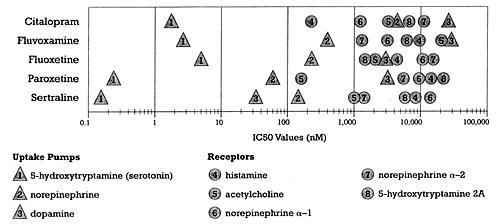 | Figure 1 - Relative potency for different sites of action for the SSRI class of antidepressants
- Based on data from Hytell 1993 |
Using 1970s technology, each SSRI was selected for development because it potently inhibited serotonin uptake without affecting any of the other neural mechanisms that plagued the use of the TCAs (Figure 1). This is why the SSRIs have the following features:
- On average, a flat dose-antidepressant response curve
- The ability to be started at an effective dose without the need to titrate
- A common adverse-effect profile
- A shared risk of causing the serotonin syndrome when used together with other types of serotonin agonists (e.g., MAOIs, certain opiates)
However, the way that the SSRIs differ from one another is directly related to the human genome project and the way that drug development has progressed in the years since they were marketed. In fact, it is doubtful that some of the currently marketed SSRIs would make it out of preclinical development today--much less be approved--given the advances in drug development over the last decade.
The following principles may help readers understand the rest of this column and will set the stage for the columns to follow:
- The goal of drug development is to reduce uncertainty about the potential action of a drug.
- The goal of rational drug development is to produce drugs that affect only the target needed for efficacy and to avoid targets that can mediate adverse consequences.
- The approval of a drug for marketing by the Food and Drug Administration (FDA) is dependent on an assessment of its benefit/risk ratio.
- The accomplishment of these goals is always limited by what is known at the time the drug is developed and approved. The last principle is another way of saying that you cannot avoid hitting something if you do not know it is out there, a la Titanic.3
For this reason, drugs are on the market that would likely not be approved if their new drug application (NDA) were submitted now. For example, because of their narrow therapeutic index, the TCAs would probably have a difficult time being marketed today. The reader might then ask why such drugs are not removed from the market. Of course, drugs are removed from time to time. Recent examples include
- terfenadine (Seldane)
- astemizole (Hismanal)
- cisapride (Propulsid)
- milbefradil (Prosicor)
However, all of those drugs were voluntarily removed by their sponsors rather than being forcibly removed by the FDA. Drugs are removed for cause when the risk associated with their use clearly exceeds the benefit. Also, the responsibility for the burden of proof is reversed in drug removal versus approval. When an NDA is submitted to the Food and Drug Administration (FDA), the burden of proof is on the sponsor to demonstrate that the drug is safe. In contrast, when the issue is FDA-directed removal from the market, the burden of proof is on the FDA to demonstrate that the risk of the drug exceeds its benefit.
Relative to the last point, consider the current issue concerning QTc prolongation. A drug such as sertindole was not able to clear the approval hurdle due to what, in the past, would have been considered a trivial QTc prolongation. However, the NDA for sertindole was submitted after the "approval bar" had been raised due to deaths resulting from terfenadine-induced Toursades de Pointes.
Now consider the TCA, amitriptyline. This antidepressant is still on the market even though for over a quarter of a century it has been known to cause death as a result of delaying intracardiac conduction .4,5 In fact, 1000-1500 deaths due to TCA poisoning were reported to the American Association of Poison Control Centers during the period from 1987 to 1996.3 That is many more deaths than ever occurred as a result of terfenadine. As I discussed in an earlier column, deaths associated with TCAs can even occur during routine practice as a result of either slow metabolism or the use of an unusually high daily dose of the medication.7 Thus, the fact that amitriptyline is on the market but sertindole is not illustrates that drug approval is an evolving process that changes as our uncertainty concerning drug action is reduced as a result of advancing knowledge.
So what does the preceding discussion have to do with the Human Genome Project, the SSRIs, and the implications for future drug development in psychiatry? It is estimated that there are 100,000 genes contained in the human genome. Of these, only 10% have been identified. With the sequencing of a human genome, the stage is set to decipher the other 90% of the genes. Each gene codes for a protein. These proteins may be structural (e.g., collagen) or functional (e.g., an enzyme). Functional proteins have primarily, if not exclusively, been the targets for drug development. Receptors are a class of functional proteins that have been useful targets for drug development in psychiatry. For any receptor, three classes of drugs can be developed:
- Agonists
- Antagonists
- inverse agonists.
An agonist acts like the endogenous neurotransmitter to turn on the receptor. An antagonist puts the receptor in neutral. An inverse agonist shifts the receptor in the opposite direction. Parenthetically, binding affinity does not tell you how the drug acts at the receptor but only its relative ability to bind to the receptor.
With the complete decoding of all the genes that make up a human, medicinal chemists will have a plethora of potential targets for drug development. This is particularly relevant to psychiatry because it is estimated that 5,000 human genes code for brain specific proteins, which creates the potential for 15,000 different classes of brain specific drugs. Thus, Bill Gates was almost undoubtedly correct when he said that computational biology and informatics will dominate the next 100 years.
Recall that the effect of any drug is determined by the following equation:
Effect = potency at x drug x biological
site of action concentration variance
The human genome project is relevant to all three of the variables in this equation. It can yield new targets for drug action. It can help researchers decipher the relevant mechanisms that determine drug concentration relative to the dose taken. And, third, it can help us determine why some patients respond to a given dose of a given drug while others do not and still others become toxic.8
So How Do the SSRIs Fit into This History?
SSRIs were developed prior to the identification of specific cytochrome P450 (CYP) enzymes. In fact, CYP 2D6 was the first CYP enzyme to have its gene isolated and identified in 1988.9 That was almost a decade after the last SSRI was synthesized and ironically the same year that the first SSRI, fluoxetine, was marketed in the United States. These facts illustrate the principle that you cannot avoid hitting what you cannot see. Parenthetically, CYP enzymes are not the only meaningful functional proteins in the human body, but they are important with regard to determining certain drug effects and even more important in terms of this discussion as illustrations of fundamental principles relevant to drug discovery and medicine.
In the 1970s, we knew that most drugs underwent biotransformation as a necessary step in their clearance from the body. We also knew that this process was principally carried out by the cytochrome P450 (CYP) system. Yet, we knew little about this system. In fact, discussions in the 1970s centered on whether there were one or two CYP enzymes. We now know there are many, many more and have developed a way of classifying them based on their sequence homology.10
We also now know that some drugs can induce or inhibit these enzymes as well as be metabolized by them. We further know that such induction or inhibition sets the stage for predictable, and thus avoidable, drug-drug interactions. In fact, this phenomenon is what led to the removal of terfenadine (Seldane), astemizole (Hismanal), cisapride (Propulsid), and milbefradil (Prosicor) from the market. Terfenadine, astemizole, and cisapride are all substrates for CYP 3A3/4, while milbefradil is an inhibitor of this enzyme.11 In the case of terfenadine, astemizole, and cisapride, the problem is that unusually high levels of these drugs can occur as a result of co-administration with a potent CYP 3A3/4 inhibitor. When the levels of these drugs become high enough, they can inhibit ion channels in the heart and thus cause fatal arrhythmias. This fact was not detected in clinical trials with these drugs because such levels do not occur in human beings unless they are also taking a potent CYP 3A3/4 inhibitor.
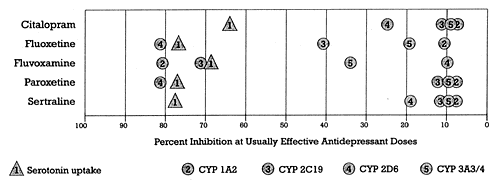 | Figure 2 - In vivo profile of SSRIs: Serotonin uptake inhibition versus CYP enzyme inhibition.
- Based on data from Shad and Preskorn15 |
Milbefradil, a T-selective calcium channel blocker, substantially inhibits CYP 3A3/4.12 As a result of this unplanned action, co-administration of milbefradil could result in substantial elevations of CYP 3A3/4 substrates/drugs such as the lipid lowering statins. At sufficiently high levels, these statins can in turn cause rhabdomyolysis.
As a result of the problems encountered with terfenadine and milbefradil, all major pharmaceutical companies now screen their new candidate drugs for effects on CYP enzymes prior to taking them into development. The goal is to determine whether the new candidate is likely to cause drug-drug interactions via effects on CYP enzymes. In fact, the substantial risk of causing this type of drug-drug interaction is generally the proverbial "kiss of death" for a new candidate drug. Upper management in most companies will not allow such candidates to go into development.13 Even if they did, it is unlikely that the FDA today would approve a drug that causes substantial inhibition of a GYP enzyme unless that drug has unique efficacy for a serious condition.14
Since the SSRIs were developed before the genes coding for CYP enzymes had been identified, they could not be screened against such targets. Figure 2 shows the relative ability of different SSRIs to inhibit the five major CYP enzymes in relation to their ability to block serotonin uptake. The latter mechanism is the one most likely responsible for their antidepressant activity.
Note that Figure 2 is not theoretical nor is it based on in vitro data. This figure shows the in vivo inhibition of these mechanisms as determined in humans under the usual dosing conditions for these drugs for antidepressant efficacy.
Those interested in how this figure was constructed are referred to the following sources. Information on the serotonin uptake inhibition is taken from data presented in my column "Finding the Signal through the Noise."16 Data on CYP enzyme inhibition is based on a recent review of in vivo studies with these drugs. This review was first presented as an invited address to the American Association of Pharmaceutical Scientists and subsequently published as a chapter in the book Metabolic Drug Interactions.15 Those studies provided data about what degree of increase in a co-prescribed drug/model substrate for a specific CYP enzyme occurs as a result of co-administration of a specific SSRI. The degree of increase is in turn a measure of the degree of inhibition of that specific CYP enzyme that must occur as a result of the SSRI. Using the mathematical formula that relates enzyme inhibition to substrate accumulation, I calculated the percentage of CYP enzyme inhibition that must have occurred. For a more detailed mathematical discussion of these calculations, the reader is referred to the appendix in my pocketbook Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors.17
All of the SSRIs are selective in terms of their effects on the neural targets shown in Figure 1. However, Figure 2 shows an important way in which the SSRIs differ. Fluoxetine, fluvoxamine, and paroxetine are all more potent as CYP enzyme inhibitors than they are as serotonin reuptake inhibitors. These three are selective for serotonin uptake inhibition relative to the other targets shown in Figure 1 but are not selective in terms of avoiding effects on CYP enzymes. In other words, the structure of these three SSRIs conveys the ability to block both the serotonin uptake pump (i.e., the desired target) as well as one or more specific CYP enzymes (i.e., unintended and undesired targets at least for antidepressants). If these drugs were in development today, medicinal chemists in the drug discovery arm of the company would be charged with using these results to modify the structure of these three SSRIs to come up with a new lead compound that affects the desired target without affecting these CYP enzymes. They would be asked to do so because a drug that affects a CYP enzyme as potently as it does its desired target will cause specific pharmacokinetic interactions when used together with drugs that are substrates for those specific CYP enzymes. In other words, these three currently marketed SSRIs might well not make it out of drug discovery today given current knowledge and technology, but instead would be used to develop still more selective lead compounds.
In this regard, SSRIs are simply an interesting example of the rapid development that has occurred in the ability to refine structure-activity relationships to develop compounds that are truly selective. The human genome project gives us more targets to hit and more targets to avoid, which is both a blessing and a curse for drug discovery.
An examination of Figures 1 and 2 also illustrates why it is important to know how the phrase "drug class" is being defined. As shown in Figure 1, all marketed SSRIs belong to the same class in terms of selective serotonin uptake inhibition. However, as shown in Figure 2, they are not all in the same class in terms of being CYP enzyme inhibitors. In fact, some SSRIs do not inhibit any CYP enzyme to a substantial degree under usual dosing conditions, whereas others inhibit one or more CYP enzymes, and not necessarily the same ones. For example, fluvoxamine does not inhibit CYP 2D6 like fluoxetine and paroxetine even though it is a substantial inhibitor of CYP 1A2 and 2C19. Thus, fluoxetine, fluvoxamine, and paroxetine could also be classified as specific CYP enzyme inhibitors. Such a grouping would include drugs that share this action. Note that not all drugs subsumed under a classification based on specific CYP enzyme inhibition would be antidepressants, since the ability to inhibit a CYP enzyme does not, as far as we know, convey antidepressant efficacy. For example, quinidine shares with fluoxetine and paroxetine the ability to cause substantial CYP 2D6 inhibition but is not an antidepressant.
Today, almost every major pharmaceutical company has the ability to screen 500,000 to 1,000,000 compounds against known biologically important human proteins in a period of approximately 2 months as a result of high-through-put screening. The results of such screening provide medicinal chemists with a considerable amount of data which can be used to develop refined structure-activity relationships, which in turn can be used to synthesize lead compounds with a high affinity for the desired target and a low affinity for nondesired targets. The concept of a lead compound will be discussed later in this series of columns.
In this regard, recall that only an estimated 10% of all of the genes coding for biologically relevant proteins in humans have so far been isolated and identified. This situation will change as a result of the human genome project and so will drug discovery, drug approval, and eventually clinical practice. In subsequent columns, I will discuss what is being done to identify novel proteins and to determine whether they are likely to be clinically useful targets for drug development.
References
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Hyttel J, Comparative pharmacology of selective serotonin reuptake inhibitors (SSRIs). Nord J Psychiatry. 1993;47 (suppl 30):5-12
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Elonen E, Mattila M, Saarnivaara L, Cardiovascular effects of amitriptyline, nortriptyline, protriptyline, and doxepine in conscious rabbits. Eur J Pharmacol 1974;28:178-88
- Preskorn SH, Irwin HA, Toxicity of tricyclic antidepressants-kinetics, mechanism, intervention: a review. J Clin Psychiatry. 1982;43:151-156
- Barbey JT, Roose SP, SSRI safety in overdose. J Clin Psychiatry 1998;59:42-8
- Preskorn SH, What happened to Tommy? J Pract Psychiatry Behav Health 1996;1:39-43
- Preskorn SH, Why did Terry fall off the dose-response curve? J Pract Psychiatry Behav Health 1998;4:363-7
- Gonzalez FJ, Skoda RC, Kimura S, et al, Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442-446
- Nebert DW, Adesnik M, Coon MJ, et al, The P450 gene superfamily: recommended nomenclature. DNA. 1987;6:1-11
- Shen DD, Madani S, Banfield C, Clement RP, H1-receptor antagonists. In: Levy RH, Thummel KE, Trager WF, Hansten P, Eichelbaum M, eds. Metabolic drug interactions. Philadelphia: Lippincott, Williams & Wilkins; 2000:435-46
- Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, eds, Metabolic drug interactions. Philadelphia: Uppincott, Williams & Wilkins, 2000
- Wrighton SA, Thummel KE, CYP3A. In: Levy RH, Thummel KE, Trager WF, Hansten PD, Eichelbaum M, eds. Metabolic drug interactions. Philadelphia: Lippincott, Williams & Wilkins; 2000: 115-34
- Williams R, Commentary on an integrated approach to the evaluation of clinically significant drug-drug interactions. American Society for Clinical Pharmacology and Therapeutics (ASCPT) March 15-17, 2000: (Abstract)
- Rodriguez AD, Evaluating drug-drug interactions: A report from the frontlines. Lecture given at ASCPT; March 15-17, 2000
- Chiba K, Kobayashi K, Antidepressants. In: Levy R, Thummel K, Trager R; et al. (eds) Metabolic drug interactions. Philadelphia: Lippincott William & Wilkins 2000:233-243
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
|



