|
|
| Relating Clinical Trials to Psychiatric Practice: Part I: The Case of a 13-Year Old on Aripiprazole and Fluoxetine |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice, July 2003, 307-313
|
Since 1990, psychiatrists and their patients have been blessed with an ever-expanding array of psychopharmaceuticals to treat a wide variety of psychiatric maladies. Nevertheless, this blessing can pose a serious challenge for practitioners as they try to keep abreast of new developments, since they have more options, each with different pharmacodynamics and pharmacokinetics, to understand and weigh. Practitioners are also faced with relating results of registration trials of new drugs to the patients they see in their day-to-day practices. This process is fraught with problems, particularly since the "usual" patient in these trials is quite different from the usual patient seen in psychiatric practice. This two-part series of columns will explore this issue. This column (Part I) presents a case that illustrates some of the difficulties that arise in extrapolating from the results of registration trials to the real world of practice. Part II will expand the discussion by considering common differences between the "usual" patient in registration trials and the typical patient in psychiatric practice. As in past case-based columns, a number of questions are posed at the end of the case presentation for the reader to consider before moving to the discussion.
THE CASE
The patient was a 13-year-old boy admitted to the hospital following an alleged suicide attempt. This admission was his third over the past year.
The patient’s psychiatric illness began at puberty about 1.5 year earlier. His parents first noticed a change in behavior when the patient became oppositional and defiant; however, he exhibited no frank antisocial behavior. The patient then became more withdrawn, although he continued to do well in school, maintaining close to an A average. His first psychiatric hospitalization occurred at age 12 and was occasioned by reports of persecutory hallucinations and delusions. His second hospitalization approximately 7 months later involved transfer to a facility several hundred miles away from home for extended care. He was treated at that facility for 3 months before returning home approximately 3 weeks prior to his suicide attempt and this admission.
His diagnosis upon discharge from his second inpatient admission was schizoaffective disorder; his medication regimen at the time of that discharge was aripiprazole, 60 mg/day, and fluoxetine, 60 mg/day. He was on this regimen at the time of this admission.
There was no family history of schizophrenia or other psychotic illness but there may have been some family members with undiagnosed bipolar type II illness. There was no personal or family history of substance abuse including alcohol abuse.
At the time of this admission, the patient’s general physical examination and laboratory tests were unremarkable. The patient’s neurological examination revealed no localizing signs, no primitive reflexes, no cerebellar findings, and no abnormal motor findings such as tremors, diminished reflexes, or myoclonus.
Mental status examination (MSE) revealed the following. The patient was oriented to person, place, time, and situation. He stated that his mood was "fine." His affect was blunted and vacuous. He was sluggish (i.e., torporous) and appeared to have diminished intensity of sensation (i.e., hypoacusis) but he was not sedated. His speech was vague, digressive, and guarded with marked latency of response and cognitive slowing but no derailment of thought. He described persecutory auditory hallucinations, visual illusions, and multiple, nonsystematized or noninetegrated bizarre delusions principally involving his family members.
Questions
- What are the most salient features of the MSE in this case?
- What diagnoses would you consider for this patient?
- Are there special laboratory tests that you would order in addition to a complete blood count, a metabolic panel, liver function tests, and a urinalysis including a screen for drugs of abuse?
- Would you continue the current treatment regimen or modify it?
- How does this case illustrate the general problem that the usual patient in many registration trials is not the typical patient seen in practice?
CASE DISCUSSION
The answer to question #1 is admittedly subjective. Certainly, this patient was hospitalized because of his alleged suicide attempt and psychotic symptoms. However, the most salient MSE features may actually be his torpor, hypoacusis, and marked cognitive slowing. In the absence of any abnormal physical and general laboratory findings, this patient’s medication regimen had to be considered as a potential cause of or contributing factor to these findings.
As with most of the cases presented in this column, the following two equations are relevant to a discussion of the medication regimen employed in this case:
Effect = affinity for/intrinsic activity at site of action
x concentration at site of action
x biological variance among patients
(Equation 1)
Drug concentration = dosing rate / clearance
(Equation 2)
The last variable in equation 1 (i.e., biological variance) is summarized by the mnemonic, GADE, which stands for Genetics, Age, Disease, and Environment. As with every patient, all of these variables must be considered in this case.
At the time of this admission, the patient was on a relatively new psychopharmaceutical, aripiprazole, a partial agonist at the dopamine-2 (D-2) receptor.1,2 As with most recently released agents, the clinical trial experience at the time of this case was limited to adults--but this patient was 13 years old. The recommended maximum dose in adults is 30 mg/day. This child was on 60 mg/day. Conceivably, children in general or this child in particular may have had rapid clearance of this drug, necessitating the use of an unusually high dose. However, there was no empirical evidence to support rapid clearance in this child. Instead, the available data indicated that the dose had been increased using the time-honored approach of dose titration based on clinical assessment of response. In this approach, the dose is advanced when there is absence of efficacy and no apparent adverse effects, as discussed in a previous column.3
Of note, the registration trial data with aripiprazole are consistent with a flat dose-response curve from 10 to 30 mg/day.1 In other words, there is on average no apparent advantage in using doses higher than 10 mg/day. A flat dose-response curve is also consistent with the apparent mechanism of action of aripiprazole, partial agonism of the D-2 receptor. With a partial agonist, once the receptors are fully or nearly fully occupied, increasing the dose will not change the level of D-2 activity. Instead, saturation of the receptor with the partial agonist can only decrease the activity of the receptor to the point where the intrinsic activity of the partial agonist is reached--in contrast to the level of zero activity that could theoretically occur when the receptor is saturated with a full antagonist.
There is, however, a caveat: flat dose-response curves are relevant to populations of patients and not to a specific patient. Registration trials tell us what occurred in the usual patient in the trials that led to approval, whereas clinicians are interested in what specifically will occur or is needed by their patient. That is the reason why clinicians find that dose titration (either up or down from what was usually found to be effective in the registration trials) can sometimes improve the response of their specific patient to a specific drug. In other words, their patient may be an outlier on the "usual" dose-response curve.4 That appeared to be the approach used in this case and the reason why this patient ended up on a dose of 60 mg/day of aripiprazole (i.e., double the maximum recommended dose for an adult patient). However, the functional dose of aripiprazole in this case may have been considerably higher because of the coprescription of 60 mg/day of fluoxetine.
Why?
To understand the last statement, it is important to know that the clearance of aripiprazole (see Equation 2 above) is principally determined by the functional activity of the drug metabolizing cytochrome P450 (CYP) enzymes 2D6 and 3A3/4, with the former usually being somewhat more important in most people based on both in vitro and in vivo human drug metabolism studies. These studies included pharmacokinetic drug-drug interaction studies done during development in which aripiprazole was co-administered with either quinidine or ketoconazole (substantial and selective inhibitors of CYP 2D6 and 3A, respectively). Co-administration of quinidine (166 mg/day for 13 days) caused a 112% increase in plasma levels of aripiprazole, whereas coadministration of ketoconazole (200 mg/day for 14 days) caused a 63% increase, when measured using the area under the curve (AUC) method.5 (The AUC method was discussed in a recent column.6)
| Figure 1 - Estimation of the relative inhibitory effect of fluoxetine on CYP 2D6, the serotonin uptake pump, and CYP 3A |  |
While quinidine and ketoconazole are selective inhibitors of CYP 2D6 and 3A, respectively, fluoxetine is a substantial but nonselective inhibitor of several CYP enzymes. As discussed in another recent column,7 20 and 40 mg/day of fluoxetine, respectively, convert 66% and 95% of individuals with normal CYP 2D6 activity into phenocopies of genetic deficiency of this enzyme. While fluoxetine is most potent at inhibiting CYP 2D6, it also inhibits CYP 2C9/10 > CYP 2C19 > CYP 3A in a concentration-dependent manner. Figure 1 illustrates the concentration-dependent effects of fluoxetine on three mechanisms of action relevant to this case: CYP 2D6, the serotonin uptake pump (i.e., the presumed mechanism responsible for its antidepressant efficacy), and CYP 3A. No studies have even been done to assess the effects of 60 mg/day of fluoxetine on these three mechanisms of action under steady-state conditions. Therefore, Figure 1 is by necessity based on an extrapolation from available data. At 20 mg/day, fluoxetine produces only modest inhibition of CYP 3A (approximately 25% decrease in the level of a model substrate such as alprazolam based on in vivo studies in humans).8 If one conservatively assumes a linear increase in inhibition with increasing dose, then 60 mg/day of fluoxetine would be expected to produce 75% increase in the AUC of a CYP 3A substrate. As discussed later, this child had a somewhat greater accumulation of fluoxetine and norfluoxetine than would be predicted based on multiplying the plasma level achieved on 20 mg/day by 3 (i.e., a linear extrapolation) consistent with fluoxetine slowing its own clearance and thus leading to a greater than dose proportional increased accumulation.
Based on the findings with quinidine and ketoconazole, the aripiprazole package insert advises that its dose should be reduced by one-half when used with either a selective CYP 2D6 or CYP 3A inhibitor alone. This child was on a dose of fluoxetine that should have produced simultaneous and pharmacologically meaningful inhibition of both CYP 2D6 and 3A (Figure 2). The effect of simultaneous inhibition of both of these drug-metabolizing enzymes on the clearance of aripiprazole has not been studied in adults or children. A conservative view is that this patient’s functional dose of aripiprazole was 120 mg/day or higher and thus at least four times higher than the recommended dose in adults. There is also little or no empirical information about the effects of such a dose of aripiprazole in adults or children.
| Figure 2 - Relationship between aripiprazole dosing rate, level, CYP 2D6 and 3A mediated clearance, and the inhibitory effect of fluoxetine on these two CYP enzymes | 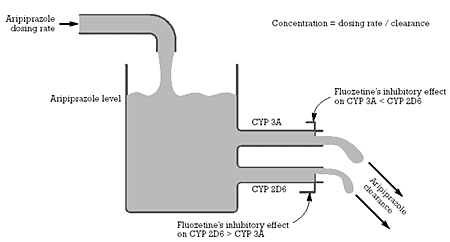 |
As is apparent based on Figure 1, fluoxetine can be properly classified as a CYP 2D6 inhibitor (i.e., a mechanism of action mediating a specific type of drug-drug interaction) as well as a serotonin uptake inhibitor (i.e., a mechanism of action predicting specific pharmacodynamic effects including antidepressant efficacy). There is no way to use fluoxetine as an antidepressant without also inhibiting CYP 2D6 because fluoxetine is as potent, or perhaps slightly more potent, an inhibitor of the human drug metabolizing enzyme CYP 2D6 as of serotonin uptake inhibition. Figure 1 also illustrates that the magnitude of the fluoxetine-induced reduction in the clearance of co-prescribed drugs dependent on one or more of the CYP enzymes inhibited by fluoxetine will be a function of the concentration of fluoxetine plus its active metabolite, norfluoxetine (which is relatively comparable to fluoxetine in terms of potency as an inhibitor at all of the sites of action in Figure 1).
The following explanation may help clarify the situation. In CYP-enzyme-mediated drug-drug interactions, the relevant site of action (variable 1 in Equation 1) is the CYP enzyme that is being inhibited by the perpetrator drug (in this case, fluoxetine). The magnitude of the reduction in enzyme activity (i.e., the effect of fluoxetine as a CYP enzyme inhibitor in Equation 1) is a function of the potency of the inhibitor (or perpetrator) (in this case, fluoxetine) and its intrinsic activity at the site of action (in this case, CYP 2D6 and CYP 3A) x the concentration of the perpetrator (which is dependent on the dosing rate of the perpetrator divided by its clearance) x any relevant biological variability in the specific patient (e.g., the clearance of fluoxetine in this 13 year old). Sources of such variability could include his genetics (e.g., family history suggestive of bipolar type II illness), age (13 with the attendant neurodevelopmental effects of "raging" sex hormones), his disease (historically, schizoaffective disorder), and his environment (e.g., fluoxetine which is inhibiting the two CYP enzymes most responsible for the biotransformation and eventual elimination of aripiprazole).
Was the fluoxetine level truly high in this case?
Equation 2 shows that the dosing rate is only one of the two variables that determine the concentration of a drug. The dose of fluoxetine in this case was also likely raised based on clinical assessment of response (i.e., inadequate efficacy and no apparent problems with adverse effects). Ironically, like aripiprazole, fluoxetine at doses of 20-60 mg/day was shown to have a flat dose-response curve in the registration trials that led to its approval as an antidepressant. One possible explanation for the increase in the fluoxetine dose is that this patient needed a higher than usual dose (due to unusually rapid ability to clear fluoxetine) to reach the levels usually achieved by the patients who received 20 mg/day during the registration trials with this drug. To assess this possible explanation for the high dose, a fluoxetine level was ordered: the levels of fluoxetine, norfluoxetine, and the combination of the two were 482 ng/ml, 214 ng/ml, and 696 ng/ml, respectively.
Many readers of this column may not be used to interpreting plasma levels of fluoxetine and norfluoxetine. Of note, the levels in this patient were measured 24 hours after the last dose of fluoxetine and were more than 3.5 times higher than peak levels (i.e., 4 hours after the last dose) measured in adults receiving 20 mg/day under steady-state conditions.9 Thus, this patient did not have unusually rapid clearance of fluoxetine necessitating a higher dose to achieve usually effective levels of this drug (Equation 2). In fact, the levels were somewhat higher than would have been predicted based on a linear relationship between increase in dose (i.e., 3 times higher than the usually effective dose) and levels (i.e., more than 3.5 times higher). This nonlinearity is consistent with the fact that fluoxetine saturates a number of cytochrome P450 enzymes, including those responsible for its clearance, in a concentration-dependent fashion. This nonlinear pharmacokinetics is also reflected in the increased norfluoxetine to fluoxetine ratio seen in this case (i.e., 482/214 > 2) in contrast to the usual ratio of 1 seen in adults in the fluoxetine registration trials. That finding is consistent with a concentration-dependent slowing of the conversion of fluoxetine to norfluoxetine.
Was the aripiprazole level truly high in this case?
Despite the considerations discussed above, this patient could conceivably have had usually rapid clearance of aripiprazole and thus may have needed this higher dose to achieve the levels usually achieved in the registration trials which used doses ranging from 10 to 30 mg/day and which formed the basis for the approval of this drug. For this reason, an aripiprazole level was also ordered. Unfortunately, no commercial laboratory could be found that does aripiprazole levels, based on calls to the local hospital laboratory, to several national reference laboratories, to Bristol-Myers Squibb, which co-markets the drug with its manufacturer, Otsuka, and to several research laboratories around the country. Thus, the level of aripiprazole in this case could not be assessed.
Certainly, the technology exists to quantify the plasma levels of aripiprazole. In fact, these levels were measured during the development of this drug, which is a standard aspect of the drug development process.10 Yet, they were not available to help guide the treatment of this child. It is interesting that a drug can be marketed without therapeutic drug monitoring (TDM) being available to help clinicians assess whether the usual dose that was found to be safe, well-tolerated, and effective in the registration trials is an appropriate dose for a specific patient in practice, given the frequently substantial differences in clearance that can be caused by genetic polymorphisms, age-related changes, or the effects of intercurrent disease or the co-ingestion of other substances (e.g., fluoxetine in this case). While there are many limitations to the interpretation of TDM results in psychiatry, it does provide quantitative data in a discipline that is otherwise heavily dependent on clinical assessment for both diagnosis and treatment monitoring.
Are there other considerations in this case?
The focus so far has been on the pharmacokinetic interaction between aripiprazole and fluoxetine--but these drugs also have the potential to interact pharmacodynamically. Fluoxetine at the concentrations measured in this child should have produced virtually complete inhibition of the serotonin uptake pump (Figure 1). The most relevant sites of action of aripiprazole (and its affinity constants [Ki’s] for these respective sites) include D2 and D3 (0.34 and 0.8 nM, respectively) and serotonin 5-HT1A and 5-HT2A (1.7 and 3.4 nM, respectively].1,2,5 Given these Ki’s, a curve similar to Figure 1 could be generated for the concentration-dependent effects of aripiprazole on these four neural sites of action (variable 1 in Equation 1). The concentration achieved in this patient may well have been beyond the concentration achieved in any patient in the formal registration trials of this drug. Certainly, aripiprazole at the concentration achieved in this child may well have completely saturated the D2 and 5-HT1A receptors, where it is a partial agonist, and have produced substantial occupation of the 5-HT2A receptor, where it is an antagonist, and of the D3 plus receptor, where its intrinsic activity (i.e., agonism, partial agonism, antagonism) has not been characterized. In fact, studies done in normal human males showed that doses of aripiprazole from 0.5 to 30 mg/day given for 14 days produced dose-dependent increases in D2 receptor occupancy that ranged from 40% to 95%.11 Since this child was on a functional dose of 120 mg/day or higher, it is likely that his D2 receptor occupancy was essentially 99%.
The combined or net effect of these individual effects of aripiprazole may never have been formally observed until this case, and certainly not in the brain of a 13-year-old child. Moreover, these effects of aripiprazole were occurring in the presence of near complete inhibition of the neuronal uptake pump for serotonin produced by the levels of fluoxetine and norfluoxetine achieved in this child. That is important because there are anatomic, physiologic, and empirical data that indicate that the dopamine and serotonin systems interact and that these specific neural mechanisms (serotonin uptake inhibition [fluoxetine] and effects on D2, D3, 5-HT1A, and 5-HT2A [aripiprazole]) interact.
An in-depth discussion of the possible consequences of affecting all of these mechanisms simultaneously is beyond the scope of this column and would be speculative at best. The following is offered simply as an illustration of the complexity of such interactions. As discussed and illustrated in a previous column,12 serotonin afferent input from the raphe nucleus inhibits dopamine cell firing in the substantia nigra. That would decrease dopamine release in the basal ganglia and could aggravate the antidopamine effect of a D2 antagonists such as perphenazine or haloperidol, resulting in severe extrapyramidal side effects or even neuroleptic malignant syndrome.12 Perhaps, fortunately for this child, he was on aripiprazole which, as a result of its partial agonist activity at D2 receptors, would prevent complete blockade of D2 activity in the basal ganglia. Beyond this brief example, further speculation on the central pharmacodynamic interactions in this case will be left to the reader.
Was there a clinically meaningful interaction?
While the discussion of the potential pharmacokinetic and even pharmacodynamic interactions in this case is interesting, the pivotal question remains: Did aripiprazole at the levels achieved in this patient, either alone or in combination with fluoxetine/norfluoxetine, contribute in any meaningful way to the MSE findings in this child? To answer that question and to treat the patient, the two drugs were discontinued. Over the course of the next week, the patient became more responsive and less torporous. His speech became more animated and latency of response diminished. However, he continued to report auditory hallucinations and delusions. Thus, aripiprazole at the levels achieved in this patient may have contributed to some of the abnormal MSE findings observed in this patient on admission. The role of fluoxetine/norfluoxetine can be ruled out to a reasonable degree since, given their long half-lives, their levels would not have been expected to change to a pharmacologically meaningful degree in 1 week (see Figure 2 in "A Message from Titanic"13).
Nevertheless, the resolution of these MSE findings might not have been due to a decrease in aripiprazole levels but instead may have been due to the natural waxing and waning of the underlying illness in this child or to the therapeutic effect of the inpatient unit and the treatment team. To address that possibility, the patient would have had to be started back on these two drugs at the same doses to see if the torpor and cognitive slowing recurred. That was not done for obvious ethical and practical reasons. The fact that such a rechallenge was not done illustrates a frequent limitation that arises when trying to establish the clinical relevance of adverse drug-drug interactions: What are the ethics of exposing patients or volunteers to a risk of toxicity to prove that the risk is real?
Even if one could demonstrate that the resolution of certain symptoms was not due simply to being on the inpatient unit, that information alone does not establish the relative contribution of aripiprazole and fluoxetine to the torpor seen in this patient. Conceivably, this effect could have been due to the pharmacodynamics of aripiprazole alone or to the combined pharmacodynamic effects of aripiprazole and fluoxetine/norfluoxetine. To answer this question, the patient would have to have been treated with both drugs together and with each drug alone at doses sufficiently high to produce the levels achieved in this patient on the combination. Again, this approach was not done for both ethical and practical reasons.
If such experiments were done with this patient, they would fall in the category of N = 1 studies in which the patient serves as his or her own control, which is in fact part and parcel of clinical practice: The patient is an N = 1 but the goal of the clinician is to help the patient, not to produce generalizable knowledge that may not benefit the patient and may even put the patient at increased risk for adverse events. That is a critical difference between clinical practice and research that aims to produce generalizable knowledge at an acceptable risk to the participant with his or her informed consent.
Of note, this case differs from most of the other drugdrug interaction cases previously discussed in this column.14 In most of these other cases, the fact that a drug-drug interaction occurred was unequivocal in part because the outcome was so dramatic--death, near death, seizures, delirium, and neuroleptic malignant syndrome. That was not the situation here. Cases such as the one described here, with all of their uncertainty, are fortunately much more the norm in practice than are drug-drug interactions presenting as serious and even life-threatening adverse events--and yet they may still be clinically important.
Registration trials and this patient
For most drugs, particularly recently marketed drugs, the clinician must rely on registration data for generalizable knowledge about what to expect from the drug in terms of relative safety, tolerability, and efficacy. However, there were several important gaps between the data available from the registration trials of aripiprazole and information needed for the treatment of this child.
First, the registration studies with aripiprazole, like those of most new drugs, were done in adults. Admittedly, there has been increased effort to provide formal data on children and adolescents as soon as possible after a new drug is released if it is likely that the drug will be used in children and adolescents. Such data will soon be forthcoming for aripiprazole.
Second, as mentioned above, the functional dose of aripiprazole used in this case was several times higher than the upper dose used in the adult registration trials. Somnolence was established as a dose-dependent effect of aripiprazole within the dose range of 10-30 mg/day used in its registration trials. It would seem prudent to anticipate that aripiprazole would also cause dose-dependent somnolence in children and adolescents until there are empirical studies in these populations to suggest otherwise. The finding of dose-dependent somnolence in adults is also consistent with the supposition that aripiprazole played a role in the torpor seen in this patient.
Third, this patient was receiving aripiprazole in the presence of high doses (and levels) of fluoxetine (plus norfluoxetine). There are no published data on such a combination. Likewise, there were probably few, if any, patients in the registration trials of aripiprazole who were simultaneously receiving any dose of aripiprazole in combination with any dose of fluoxetine, and almost certainly not 60 mg/day of both of these drugs. Thus, there are probably few, if any, human data on the safety, tolerability, pharmacokinetics, or efficacy of this combination at these doses.
SUMMARY
There are several major points raised by this column. The first is an appreciation of the complexity of the interactions that can occur when even just two drugs are used in combination. The second is an appreciation of how clinicians by necessity must frequently use medications in patients and situations that are quite different from the ones studied in the registration trials that led to drug approval. The third is the value of TDM to better define how close or how far the patient is from the concentrations that were studied in the registration trials. The fourth is the uncertainty that arises in trying to determine whether a less than optimal outcome has occurred in spite of the treatment plan or in part because of the treatment plan.
While these issues are specific to this case, they are also relevant to a general discussion of the limitations of registration trial data that clinicians must take into account when treating their patients. Part II of this column will expand this discussion and consider the issue of the "usual" patient in registration trials of psychopharmaceuticals versus the "usual" patient in psychiatric practice.
References
- McGavin JK, Goa KL, Aripiprazole. CNS Drugs 2002;16: 779-88
- Burris KD, Molski TF, Xu C, et al, Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 2002;302: 381-9
- Preskorn SH, Polypharmacy in a patient with refractory major depression: Part I. The case. Journal of Psychiatric Practice 2002;8:370-6
- Preskorn SH, Why did Terry fall off the dose-response curve? J Pract Psychiatry Behav Health 1998;4:363-7
- Aripiprazole package insert, Bristol-Myers Squibb.
- Preskorn SH, Reproducibility of the in vivo effect of the selective serotonin reuptake inhibitors on the in vivo function of cytochrome P450 2D6: An update (part I) Journal of Psychiatric Practice 9(2), March 2003
- Preskorn SH, Reproducibility of the in vivo effect of the selective serotonin reuptake inhibitors on the in vivo function of cytochrome P450 2D6: An update (part II) Journal of Psychiatric Practice 2003;9:228-36
- Chiba K, Kobayashi K, Antidepressants. In: Levy R, Thummel K, Trager R; et al. (eds) Metabolic drug interactions. Philadelphia: Lippincott William & Wilkins 2000:233-243
- Preskorn SH, Alderman J, Chung M, Harrison W, Messig M, Harris S, Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J Clin Psychopharmacol. 1994;14:90-98
- Preskorn SH, Modern drug development and the Human Genome Project. (collection of 4 columns published in the Journal of Psychiatric Practice). Accessed June 25, 2001
- Yokoi F, Grunder G, Biziere K, et al, Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): A study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 2002;27:248-59
- Preskorn SH, I don’t see ’em. J Prac Psych Behav Hlth. 1997; 3:302-307
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Preskorn SH, The polypharmacology section. (collection of columns published in the Journal of Psychiatric Practice). Accessed July 10, 2002
|



