|
|
| "I don't see 'em!" |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, September 1997, 302-307
|
I dont see ’em." That is what some physicians say about pharmacokinetically mediated drug-drug interactions. However, "not seeing" does not equal "not occurring." What makes this type of interaction problematic is that physicians can "see" consequences but not recognize the cause. To illustrate this point, in this column, I will discuss just such a case of "seeing" but not "recognizing."
Why Are Pharmacokinetic Drug-Drug Interactions "Not Seen?"
First, lets review some general background. Obviously, adverse drug-drug interactions often occur in a complex medical setting; the patient may have more than one illness or an illness that has been refractory to monodrug therapy.1 Hence, the physician must often consider multiple possible explanations for the adverse event, including the underlying illness, the development of a new illness, or a problem with the patients "sensitivity" or "resistance" to the treatment regimen.
In addition to these possibilities, the adverse outcome may also be due to an untoward interaction among the drugs the patient is taking. In the case of pharmacodynamic interactions, the effects of one drug on its site(s) of action may amplify or diminish the response of the patient to the effect of a coprescribed drug on its site(s) of action (e.g., a serotonin uptake inhibitor and a monoamine oxidase inhibitor). In the case of a pharmacokinetic interaction, one drug may alter the absorption, distribution, metabolism, or elimination of a coprescribed drug and hence its effective concentration at its site(s) of action (e.g., a nonsteroidal anti-inflammatory drug and lithium).
For several reasons, physicians may be more likely to consider pharmacodynamic rather than pharmacokinetic drug-drug interactions as a possible explanation when a serious adverse event or less than optimal treatment outcome occurs. First, until recently, we knew more about pharmacodynamic than pharmacokinetic drug-drug interactions. However, over the last decade, there has been an explosion in our knowledge, particularly of cytochrome P450 (CYP) interactions.2 Second, there has been some controversy about the importance of pharmacokinetic interactions.3 Third and perhaps most important, these interactions can present clinically in a myriad of ways.
Certainly, pharmacokinetic drug-drug interactions can present as dramatic, life-threatening events. However, they can also present in more mundane but nonetheless clinically important ways. Generally, these interactions cause an increase or decrease in the effective concentration of the affected drug and hence often produce the same clinical outcome that would occur if the dose of the affected drug were changed to a comparable degree. Therefore, these interactions generally present as dose-dependent rather than novel events. Examples include:
- the development of an extrapyramidal side effect on an apparently low dose of a neuroleptic due to a reduction in clearance
- bradycardia on an apparently low dose of a beta blocker due to a reduction in clearance
- a breakthrough seizure in a patient on previously adequate anticonvulsant therapy due to an increase in clearance
- the development of withdrawal symptoms in a patient who has been on a stable dose of a benzodiazepine for several months or more due to an increase in clearance.
All these events can and have occurred as the result of adding or discontinuing a drug that altered the pharmacokinetics (e.g., clearance in these cases) and hence the functional dose of the affected drug.
In such instances, physicians may "see" the undesired clinical outcome but not recognize the cause. Instead, they may attribute the outcome to the patient either being "sensitive" to a known dose-dependent adverse action of the affected drug or being "resistant" to the benefits of the affected drug. What they do not realize is that the functional dose is not the prescribed (or apparent dose), because of an alteration in the rate of drug clearance caused by the co-prescribed drug.
As a result of not "seeing" the etiology of the problem, the physician may take one of several courses of action: stop the affected drug, add an antidote (e.g., adding an anticholinergic to a neuroleptic), or ironically even increase the dose of the affected drug. The last can occur when drug toxicity is misinterpreted as a worsening of the underlying illness.4
The need for an awareness of these interactions is underscored by the fact that a sizable percentage of patients, perhaps the majority in many clinical settings, are taking multiple medications.5, 6 In fact, the need to be knowledgeable about drug-drug interactions is one of the most important reasons why extensive training and experience is needed to safely and effectively prescribe medications.
A Case of Neuroleptic Malignant Syndrome
Let us now review a case in which untoward consequences were observed and even published but the cause was not recognized.7 The fact that the author, the external reviewers, and an editor did not apparently consider the role of drug-drug interactions in the etiology of the neuroleptic malignant syndrome (NMS) underscores that "not seeing" is not the same as of "not occurring." This case also illustrates that pharmacodynamic and pharmacokinetic drug-drug interactions are not mutually exclusive; rather there can be a complex interplay between these two types of drug-drug interactions, in which each amplifies the others clinical consequences.
My comments in this column are not meant to detract in any way from the author of the case report, the reviewers, the editor, or the journal. The author was obviously a concerned and thoughtful physician who took the time and trouble to publish this case. The journal is one of the premier journals of its type and has done much to promote the safe and effective use of psychotropic medications.
This discussion should also not be construed as a blanket indictment of the use of polypharmacy. Physicians generally have good reasons to use more than one medication in combination.1 For many patients, polypharmacy is a necessity--to do otherwise would be inappropriate care. In fact, physicians frequently use more than one medication in combination to produce a therapeutic drug-drug interaction in which the second drug is added to either increase or reduce some specific effect of the primary medication. In this column, however, I focus on the problem that arises when such interactions occur unintentionally and the cause is not recognized, even though the consequence is "seen."
As I review this case, you may wish to test your pharmacological detective skills by taking a moment to jot down your explanation after the facts of the case are presented. For those less inclined to detective games, you may wish to refer to Figure 1 at the end of the column in advance and then as needed as the discussion unfolds.
The case involved a 48-year-old woman with bipolar disorder who had been on a regimen consisting of paroxetine (20 mg/day), lithium carbonate (1200 mg/day), and trazodone (200 mg at night) for an unspecified period of time. Perphenazine (16 mg/day) and benztropine (2 mg/day) were added to this regimen to treat hypomanic symptoms. Two weeks later, the patient was hospitalized for NMS (i.e., altered consciousness, lead pipe rigidity, myoclonic twitching, hyperthermia, leukocytosis, and elevated serum creatinine kinase).
Why Did NMS Occur?
Why did NMS occur? (For those so inclined, now is the time to formulate your explanation.) Was the patient sensitive to perphenazine? NMS is so named because it is generally caused by neuroleptic medications.
Did lithium play a role? There have been reports suggesting that increased neurotoxicity may result from the combined use of lithium and neuroleptics, particularly haloperidol.8
In his discussion, the author of the case study focused on the possible role of serotonin (SE or 5-HT), noting the similarities between NMS and the serotonin syndrome and the fact that SE has an inhibitory effect on the release of dopamine from the substantia nigra.9 He also commented on the possible contribution of meta-chlorophenylpiperazine, a metabolite of trazodone and a 5-HT2B/2C agonist.10, 11
Notice that all these possibilities involve pharmacodynamic explanations. Without a doubt, NMS or any other effect will not occur unless an appropriate mechanism of action is engaged. Nevertheless, the magnitude of the effect is also a function of the extent to which the mechanism is engaged, and that is determined by pharmacokinetics.
| (Equation 1) | | Effect | = pharmacodynamics | | x pharmacokinetics | | x biology of the patient |
As indicated in Equation 1,12 the magnitude of the effect is a function of the potency of the drug at its site(s) of action times its concentration at the site of action. Potency is the percentage of the maximum possible effect of a drug achieved as a function of its concentration. However, the absolute magnitude of the effect is determined by potency (% effect/concentration) times concentration. For example, one molecule of dopamine (DA) antagonist, regardless of its potency, will block only one receptor, which is not sufficient to cause any discernible effect, much less NMS.
Equation 1 can also be applied when using more than one medication to treat a patient and can help us conceptualize the answers to the following questions: Is there any physiological interplay between the various site(s) of action affected by the different medications the patient is taking? Does one drug alter the effective concentration of a coprescribed drug at its site(s) of action? As equation 1 makes clear, pharmacodynamic and pharmacokinetic interactions can occur simultaneously in the same patient. That was what happened in this case. First there was the inhibition of a CYP enzyme which reduced the clearance of several of the agents this patient was taking and thus increased their effective concentration at their respective sites of action. Second, the effects of these different drugs on their sites of action interacted to cause the NMS.
CYP 2D6 Activity
Let us begin the formal discussion with a question: What do meta-chlorophenylpiperazine (mCPP), paroxetine, and perphenazine have in common? Certainly, they are all psychoactive substances; however, their mechanisms of action are quite different. Another answer is that the clearance of all three is dependent on the functional activity of the CYP enzyme, CYP 2D6 (Figure 1b).13, 14, 15 There are two predominant determinants of CYP 2D6 activity: 1) genetic polymorphisms (i.e., discrete populations with different levels of activity based on genetics),16 and 2) medications that are capable of producing substantial inhibition of this CYP enzyme under usual dosing conditions.
Several mutations of the CYP 2D6 gene exist that can result in a nonfunctional enzyme.17 The incidence of these mutations varies widely in different ethnic populations. The highest incidence is in northern Europeans: approximately 5% of these individuals are homozygous for a mutant CYP 2D6 gene and hence are CYP 2D6 deficient and unable to metabolize drugs via this pathway. Such individuals develop unusually high levels of drugs that are normally metabolized by this CYP enzyme and hence are termed "poor metabolizers" (PMs) in contrast to the normal population who are termed "extensive metabolizers" (EMs).
It is now possible to detect both EMs (95% or more of the population) and PMs (5% or less of the population) using genetic testing. In the past, such a classification was made by giving individuals a substance (e.g., dextromethorphan [DM]) that is predominantly or exclusively metabolized by CYP 2D6 and then measuring the clearance of DM or its metabolite dextrorphan (DO).18 The ratio of DM:DO in the urine can reliably distinguish EMs from PMs.
This same test can be used to assess whether a drug inhibits CYP 2D6 sufficiently to convert EMs to phenocopies of PMs. Of note, 65% of EMs are converted to phenocopies of PMs when treated with 20 mg/day of either paroxetine or fluoxetine and 95% are converted when treated with 40 mg/day of fluoxetine.19, 20, 21 This does not occur with the other serotonin selective reuptake inhibitors (SSRIs) or with venlafaxine.
Perphenazine Levels
In patients who are genotypically PMs, the plasma levels of perphenazine are four times higher than in EMs on the same dose (Table 1).15 The same should be true for the 65% of EMs who are converted to PMs by paroxetine. Even in the EMs who are not fully converted to PM status as a result of paroxetine treatment, the plasma levels of perphenazine would be substantially higher than expected based on the prescribed dose. Thus, the functional dose of perphenazine in this patient was closer to 64 mg/day than to 16 mg/day. Since the degree of DA receptor blockade is a function of the plasma level of the blocker times its concentration, the degree of DA receptor blockade in this patient should also have been approximately four times higher than the physician would normally expect for the dose prescribed. This effect alone was likely pivotal in causing NMS in this patient, but there was more (Figure 1a).
| Table 1. Case Study Medication Regimen: Apparent Daily Dose Versus Functional Daily Dose | | Medication | Apparent Dose (mg/d) | Functional Dose | | Paroxetine | 20 | Paroxetine accumulation is 10 times higher and half-life is twice as long after multiple daily doses than after a single dose | | Perphenazine | 16 | Approximately 64 mg/d | | Trazodone | 200 | Parent drug levels unchanged--but levels of mCPP increased 4-10 times | | Lithium | 1200 | Unchanged | | Benztropine | 2 | May be dependent on CYP 2D6 based on case reports |
mCPP Levels
mCPP is a psychoactive metabolite of trazodone. It is formed by CYP 3A3/4 mediated cleavage of trazodone at the C-N bridge. While its production is dependent on CYP 3A3/4, the clearance of mCPP is dependent on CYP 2D6 mediated ring hydroxylation (Figure 1b). Levels of mCPP are 4-10 times higher in PMs than in EMs.14 Fluoxetine does and paroxetine should increase mCPP levels to a similar degree due to their inhibition of CYP 2D6. That is relevant to this case because mCPP reduces the activity of DA neurons in the ventral tegmental area (VTA) and in the substantia nigra, pars compacta (SNc) by its agonistic effects on 5-HT2C/2B receptors.9, 22 Thus, the inhibition of DA release produced by mCPP causes a further functional decrease in DA activity beyond that produced by perphenazines DA receptor blockade (Figure 1a). The magnitude of these two effects would be increased, because the levels of both mCPP and perphenazine are elevated due to the decrease in their clearance caused by the paroxetine-induced inhibition of CYP 2D6 (Figure 1b).
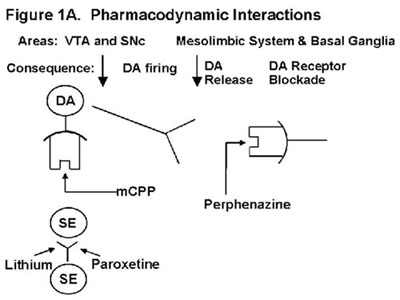 | | Legend 1A: Serotonin (SE or 5-HT) is an inhibitory afferent projection to dopamine (DA) nuclei in the ventral tegmentum area (VTA) and substantia nigra, pars compacta (SNc). Lithium, paroxetine, and mCPP increase the SE inhibitory input to VTA and SNc nuclei but through different mechanisms of action: Lithium facilitates SE release. Paroxetine blocks SE reuptake. mCPP is a 5-HT2B/2C agonist. These actions decrease DA neuronal activity and hence DA release in its terminal projections. The decreased DA release amplifies the DA deficiency produced by the perphenazine blockade of DA receptors. |
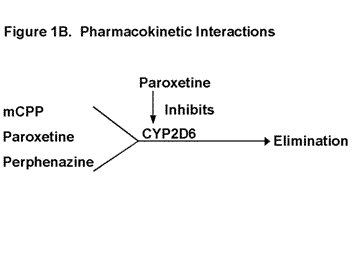 | Legend 1B: mCPP, paroxetine, and perphenazine are all dependent on CYP 2D6-mediated metabolism for their clearance. Thus the levels of all three would be increased by paroxetine inhibition of this CYP enzyme. |
Paroxetine Levels
Paroxetine is also dependent on CYP 2D6 for its metabolism at low levels but saturates this CYP enzyme at usual steady-state concentrations achieved on 20 mg/day (Figure 1b).13, 23 This saturation or autoinhibition is the basis for paroxetines nonlinear pharmacokinetics and the fact that its concentration increases 10-fold and its half-life doubles from 10 to 20 hours after repeated doses.23, 24
Consistent with the effects of mCPP on DA neurons, the VTA and SNc receive rich inhibitory SE afferent innervation from the midbrain raphe nuclei.9, 25-29 Acute administration of SSRIs (i.e., citalopram and fluoxetine) causes a dose-dependent decrease in DA neuronal activity in the VTA consistent with their ability to increase the intrasynaptic concentration of SE by inhibiting its neuronal reuptake.29 This mechanism has been proposed to account for the akathisia that can occur on SSRIs. A similar decrease in SNc activity could be responsible for the rare occurrence of Parksonism on SSRIs.
Effects of Lithium
But wait. As they say in the Vegomatic commercial, there is more. One of the many, many actions of lithium is to increase the release of SE. That was the basis for de Montingys original proposal to use lithium to augment the efficacy of tricyclic antidepressants.30 It is also the likely explanation for why SE mediated adverse effects can increase following the addition of lithium to an SSRI. In this case of NMS, the reduction of DA neuronal activity in the midbrain should have been amplified by the combined effects of lithium and paroxetine on inhibitory SE input to the VTA and SNc nuclei: lithium facilitating SE release and paroxetine slowing its clearance from the cleft (Figure 1a). These effects would add to the mCPP-induced inhibition of these neurons. Thus, lithium, mCPP, and paroxetine would all decrease the release of DA from its terminal fields, which in turn would enhance the functional deficiency of DA produced by direct DA receptor blockade caused by perphenazine (Figure 1a). The effect of each of these drugs was further magnified by their increased accumulation as a result of paroxetine-induced reduction in their CYP 2D6 mediated clearance (Table 1).
Effects of Benztropine
But wait, there was even one more player--benztropine. This agent may well have had both beneficial and deleterious consequences for this patient. Benztropine and related anticholinergic agents treat neuroleptic-induced Parkinsonism by restoring the balance between DA and ACh tone in the extrapyramidal system by blocking acetylcholine (Ach) receptors on interneurons in this system. However, one of the cardinal features of NMS is hyperthermia due to the lead pipe rigidity and myoclonic twitching. By blocking ACh receptors in the periphery, benztropine would have reduced the ability of this patient to dissipate this heat by sweating. The metabolism of benztropine has not been established; however, some authors have suggested it may be dependent on CYP 2D6 based on case reports of interactions with paroxetine.31
Conclusion
This case then is an excellent example of several clinically important points:
- pharmacokinetic and pharmacodynamic drug-drug interactions can occur in the same patient and can potentiate each other, as illustrated in Figure 1,
- physicians need to be knowledgeable about and consider the potential for both pharmacodynamic and pharmacokinetic drug-drug interactions when they treat a patient with more than one medication,
- pharmacokinetic drug-drug interactions can be misinterpreted as a "sensitivity" or "resistance" problem on the part of the patient.
This case is not unique--there have unfortunately been many other cases in which physicians saw the untoward consequences of such interactions (e.g., sudden death) but did not recognize the cause. In more than one instance, the cause was not determined until the postmortem toxicology report came back. No one knows how often such interactions have caused more subtle problems, such as an increase in nuisance adverse effects or reduced efficacy. While perhaps not medically serious, such scenarios do have health care implications in terms of optimal patient outcome and cost effectiveness. They can lead to an otherwise effective treatment being stopped or to the addition of other medications to treat the adverse effect.
A few rules of thumb can help clinicians avoid these problems:
- Consider the possibility that the patient may not be doing well because of the medications they are taking rather than in spite of the medication. When in doubt, attempt to simplify the regimen rather than simply adding more medications, particularly if the regimen is already complicated.
- Be aware of all the medications the patient is taking--not just the medications you are prescribing; this should include both over-the-counter and prescription medications.
- Consider whether the medications may interact based on their mechanisms of action or on their pharmacokinetics
- Avoid medications that affect targets (e.g., receptors, CYP enzymes) that are not essential for their therapeutic efficacy.
References
- Preskorn SH, Lacey R, Polypharmacy: when is it rational? J Prac Psych Behav Hlth. 1995;1:92-98
- Preskorn SH, What is the message in the alphabet soup of cytochrome P450 enzymes?. J Prac Psychiatry Behavioral Hlth. 1995;1:237-240
- Preskorn SH, Let the data speak to you. J Pract Psychiatry Behav Health 1997;3(1):45-8
- Preskorn SH, Do you believe in magic? J Pract Psychiatry Behav Health 1997;3:99-103
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Shad MU, Carmichael CA, Preskorn S, Horst WD, Prevalence of polypharmacy in different clinical settings and its relation to drug-drug interactions. Presented at the annual American Psychiatric Association meeting, San Diego, CA, May 17-22, 1997
- Young C, A case of neuroleptic malignant syndrome and serotonin disturbance (letter to the editor) J Clin Psychopharmacol 1997;17(1):65-6
- Janicak PG, Davis JM, Preskorn SH, Syd FJ Jr, Principles and Practices of Psychopharmacotherapy. Baltimore, Md: Williams and Wilkins; 1993
- Kelland MD, Freeman AS, Chiodo LA, Serotonergic afferent regulation of the basic physiology and pharmacological responsiveness of nigrostriatal dopamine neurons. J Pharmacol Exp Ther. 1990;253:803-811
- Curzon G, Kennett GA, mCPP: a tool for studying behavioral responses associated with 5-HT1c receptors. Trends Pharmacol Sci 1990;11:181-2
- Kennett GA, Wood MD, Glen A, et al, In vivo properties of SB 2000646A, a 5-HT2C/2B receptor antagonist. Br J Pharmacol 1994;111:797-802
- Preskorn SH, Why did Terry fall off the dose-response curve? J Pract Psychiatry Behav Health 1998;4:363-7
- Harvey AT, Preskorn SH, Cytochrome P450 enzymes: interpretation of their interactions with selective serotonin reuptake inhibitors. Part I. J Clin Psychopharmacol. 1996;16:273-285
- Barbhaiya RH, Buch AB, Greene DS, Single and multiple dose pharmacokinetics of nefazodone in subjects classified as extensive and poor metabolizers of dextromethorphan. Br J Clin Pharmacol 1996;42:573-81
- Dahl-Puustinen ML, Liden A, Alm C, Nordin C, Bertilsson L, Disposition of perphenazine is related to polymorphic debrisoquin hydroxylation in human beings. Clin Pharmacol Ther 1989;46:78-81
- Preskorn SH, Have you phenotyped your patient lately? J Pract Psychiatry Behav Health 1996;2(2):115-7
- Marez D, Legrand M, Sabbagh N, et al, Polymorphism of the cytochrome P450 CYP2D6 gene in a European population: characterization of 48 mutations and 53 alleles, their frequencies and evolution. Pharmacogenetics 1997;7:193-202
- Dayer P, Leemann T, Striberni R, Dextromethorphan O-demethylation in liver microsomes as a prototype reaction to monitor cytochrome P-450 db1 activity. Clin Pharmacol Ther 1989;45:34-40
- Brøsen K, Hansen JG, Nielsen KK, Sindrup SH, Gram LF, Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol. 1993;44:349-355
- Ereshefsky, L, Drug-drug interactions involving antidepressants: focus on venlafaxine. J Clin Psychopharmacol 1996;16(3)(Suppl 2):37-50
- Otton SV, Wu D, Joffe RT, Cheung SW, Sellers EM, Inhibition by fluoxetine of cytochrome P-450 2D6 activity. Clin Pharm Therap. 1993;53:401-409
- Prisco S, Pagannone S, Esposito E, Serotonin-dopamine interaction in the rat ventral tegmental area: an electrophysiological study in vivo. J Pharmacol Exp Ther 1994;271:83-90
- Kaye CM, Haddock RE, Langley PF, et al, A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr Scand. 1989;80(supp 350):60-75
- Alderman J, Preskorn S, Greenblatt D, et al, Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 1997;17:284-91
- Azmitia EC, Segal M, An autoradiographic analysis of the differential ascending projections of the dorsal and median raphe nuclei in the rat. J Comp Neurol 1978;179:641-68
- Mori S, Matsuura T, Takino T, Sano Y, Light and electron microscopic immunohistochemical studies of serotonin nerve fibres in the substantia nigra of the rat, cat and monkey. Anat Embryol 1987;176:13-8
- Phillipson OT, Afferent projections to the ventral tegmental area of Tsai and interfascicular nucleus: a horseradish peroxidase study in the rat. J Comp Neurol 1979;187:117-44
- Steinbusch HWM; Björklund A, Hökfelt T, Kuhar MJ, eds, Handbook of Chemical Neuroanatomy. Amsterdam: Elsevier, 1984; Serotonin-immunoreactive neurons and their projections in the CNS. Classical transmitters and transmitter receptors in the CNS. Part II. p. 68-125
- Prisco S, Esposito E, Differential effects of acute and chronic fluoxetine administration on the spontaneous activity of dopaminergic neurones in the ventral tegmental area. Brit J Pharm 1995;116:1923-31
- DeMontigny C, Cournoyer G, Morissette R, Langlois R, Caille G, Lithium carbonate addition in tricyclic antidepressant-resistant unipolar depression. Arch Gen Psychiatry 1983;40:1327-34
- Armstrong SC, Schweitzer SM, Delirium associated with paroxetine and benztropine combination. Am J Psychiatry. 1997;154:581-582. Letter
|



