|
|
Outpatient Management of Depression
6 - The Rational Basis for the Development and Use of Newer Antidepressants |
|
|
|
|
|
A decade ago, the practitioner had limited options for treating
patients with clinical depression. Those options were:
- Tricyclic antidepressants (TCAs)
- Monoamine oxidase inhibitors (MAOIs)
- Trazodone.
| TABLE 6.1 Classification
of Antidepressants By Putative Mechanism(s) of Action
Responsible for Antidepressant Efficacy:* Generic/(Trade)
Names By Drug Class |
|
Mixed Reuptake and Neuroreceptor Antagonists
Amitriptyline (Elavil)
Amoxapine (Ascendin)
Clomipramine (Anafranil)
Doxepin (Sinequan)
Imipramine (Tofranil-PM)
Trimipramine (Surmontil)
|
Norepinephrine Selective
Reuptake Inhibitors (NSRIs)
Desipramine (Norpramin)
Maprotiline (Ludiomil)
Nortriptyline (Pamelor, Aventyl)
Protriptyline (Vivactil) |
Serotonin Selective Reuptake
Inhibitors (SSRIs)
Citalopram (Celexa)
Fluoxetine (Prozac)
Fluvoxamine (Luvox)
Paroxetine (Paxil)
Sertraline (Zoloft) |
Serotonin and Norepinephrine
Reuptake Inhibitor (SNRI)§
Venlafaxine (Effexor) |
Serotonin-2A (5-HT2A)
Receptor Blocker and Weak Serotonin Uptake Inhibitor||
Nefazodone (Serzone)
Trazodone (Desyrel) |
Serotonin (5-HT2A and
2C) and a-2 Norepinephrine Receptor Blocker||
Mirtazapine (Remeron)
|
Dopamine and Norepinephrine
Reuptake Inhibitors
Bupropion (Wellbutrin, Wellbutrin SR, Zyban Sustained-Release) |
Monoamine Oxidase Inhibitors
(MAOIs)¶
Phenelzine (Nardil)
Tranylcypromine (Parnate) |
| Abbreviations: 5-HT, 5-hydroxytryptamine
(serotonin). |
* The presumptive mechanism
of action for each drug is based on the preclinical
pharmacology of the drug and the fact that it and/or
its active metabolites reach sufficient concentration
in vivo to affect this site of action, given
its in vitro potency.
All of these drugs are tertiary amine tricyclic
antidepressants (TCAs) except amoxapine.
All of these drugs are secondary amine TCAs except
maprotiline. There are nontricyclic NSRIs available
in other parts of the world, such as reboxetine. Those
drugs share the ability to inhibit norepinephrine
uptake with the secondary amine TCAs and maprotiline
but do not carry the risk of serious toxicity even
after a substantial overdose.
§ At present, venlafaxine is the only member of this
class available in the United States although several
others are in various stages of clinical testing.
|| Both nefazodone and mirtazapine also have other
mechanisms of action that are engaged at concentrations
which occur under clinically relevant dosing guidelines
(see Table 6.2).
¶ Only irreversible and nonselective monoamine oxidase
inhibitors (MAOIs) are available in the United States,
but selective and reversible MAOIs are marketed elsewhere
in the world. |
The practitioner now has 22 different antidepressants from
eight different mechanistically defined classes of antidepressants
(Table 6.1).
This chapter will explain how this mechanistically based
classification system can be used to conceptualize the clinical
relevance of the various antidepressants available to treat
patients. Thus, this chapter can be used as a reference when
reading subsequent chapters. The goal is to summarize the
knowledge necessary to:
- Predict the clinical effects of different antidepressants
- Choose a specific antidepressant for a specific patient
based on those predicted effects
- Rationally think about sequential treatment options for
the patient who has not benefited from a trial of a specific
type of antidepressant
- Anticipate potential drug-drug interactions when using
a specific antidepressant in combination with other medications.
Rational Drug Development
Drug development in psychiatry has evolved
from a process based on chance to one based on molecularly
targeting specific sites of action in the central nervous
system (CNS) (ie, specific neuroreceptors or neuronal uptake
pumps for neurotransmitters). The goal of such development
is to affect only mechanisms mediating antidepressant efficacy
while simultaneously avoiding affects on other mechanisms
which mediate tolerability and/or safety problems.181,183,236
The intent is to produce antidepressants which are:
- Safer
- Better tolerated
- Less likely to interact with other coprescribed drugs
than was the case with the older agents (ie, TCAs, MAOIs
and trazodone).
The success of this approach is underscored by the fact
that the risk of the clinical depression is now clearly worse
than the risk of the treatment such that even a hint of clinical
depression may be sufficient to warrant an empirical trial
of a newer antidepressant. That explains the substantial expansion
in the use of antidepressants that has occurred over the last
decade.164
The eight functional pharmacologic classes of antidepressants
based on their presumed mechanism(s) of antidepressant action
are listed in Table 6.1. Table 6.2 indicates which mechanisms
of action are engaged by these different antidepressants at
their usually effective antidepressant dose. In Table 6.3,
the clinical consequences that occur as a result of blocking
specific sites of action are listed. By using Tables 6.2 and
6.3 in combination, the clinician can determine what specific
effects a particular antidepressant will produce in the usual
patient on the usual antidepressant dose. Tables 6.4 through
6.7 summarize the usual adverse effects produced by these
various drugs based on the results of double-blind, placebo-controlled
clinical trials. These tables will serve as reference points
throughout the remainder of the book.
Relationship Between Pharmacodynamics,
Pharmacokinetics and Interindividual Variability
The nature and magnitude of a drug's effect is determined
by its:
- Site of action
- Binding affinity for that site
- Concentration at the site.170
These factors determine the "usual" effect of the
drug in the "usual" patient on the "usual"
dose as determined in a clinical trial. However, all patients
are not "usual" due to interindividual variability
caused by factors such as:
- Age
- Genetics
- Gender
- Intercurrent diseases affecting organ function
- Concomitant drug therapy
- Social habits (eg, smoking).131
The clinician must take into account how a specific patient
may differ from the "usual" patient in a clinical
trial when selecting and dosing a drug. The three important
variables determining the effect of a drug on a patient are
summarized in the following equation:
(Equation 1)
| Effect = |
pharmacodynamics |
x |
pharmacokinetics |
x |
interindividual variance |
This equation can be restated as follows:
(Equation 2)
| Effect = |
potency for site of
action |
x |
concentration at site
of action |
x |
interindividual variance |
The first two terms in this
equation explain the relationship between pharmacodynamics
and pharmacokinetics. The first term determines the nature
of the drug's effect and how much drug is needed at the site
of action to engage that site to a clinically meaningful extent
in the usual patient. The second term in the equation determines
the magnitude of the drug's effect by determining how much
drug reaches the site of action. The third term explains how
interindividual variability in the patient can shift the dose-response
curve (ie, greater or lesser effect than usually expected
relative to the dose prescribed).
| TABLE 6.2 Comparison
of the Mechanisms of Action of Antidepressants* |
| Mechanism of Action |
Ami-triptyline |
Desi-pramine |
Ser-traline |
Ven-lafaxine |
Nefa-zodone |
Mir-tazapine |
Bupro-pion |
Tranylcy- promine |
| Histamine-1 receptor blockade |
Yes |
No
|
No
|
No |
No |
Yes |
No |
No |
| Acetylcholine receptor blockade |
Yes |
No |
No |
No |
No |
No |
No |
No |
| NE uptake inhibition |
Yes |
Yes |
No |
Yes |
No |
No |
Yes |
No |
| 5-HT2A receptor blockade |
Yes |
No |
No |
No |
Yes |
Yes |
No |
No |
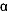 -1
NE receptor blockade -1
NE receptor blockade |
Yes |
No |
No |
No |
Yes |
No |
No |
No |
| 5-HT uptake inhibition |
Yes |
No |
Yes |
Yes |
Yes |
No |
No |
No |
| -2
NE receptor blockade |
No |
No |
No |
No |
No |
Yes |
No |
No |
| 5-HT2C receptor blockade |
No |
No |
No |
No |
No |
Yes |
No |
No |
| 5-HT3 receptor blockade |
No |
No |
No |
No |
No |
Yes |
No |
No |
| Fast Na+ channels
inhibition |
No |
No |
No |
No |
No |
No |
No |
No |
| Dopamine uptake inhibition |
No |
No |
No |
No |
No |
No |
Yes |
No |
| Monoamine oxidase inhibition |
No |
No |
No |
No |
No |
No |
No |
Yes |
| Abbreviations: NE,
norepinephrine; 5-HT, 5-hydroxytryptamine (serotonin);
Na+, sodium. |
* Amitriptyline
represents the mixed reuptake and neuroreceptor blocking
class, desipraminenorepinephrine selective reuptake inhibitors,
sertralineserotonin selective reuptake inhibitors, venlafaxineserotonin
and norepinephrine reuptake inhibitors, nefazodone5-HT2A
and weak serotonin uptake inhibitors and mirtazapinespecific
serotonin and norepinephrine receptor blockers, bupropiondopamine
and norepinephrine uptake inhibitor. Monoamine oxidase
inhibitors (MAOIs) do not directly share any mechanism
of action with other classes of antidepressants, although
they affect dopamine, norepinephrine, and serotonin neurotransmission
via their effects on monoamine oxidase.
The effects of these various antidepressants are listed
using a binary (yes/no) approach for simplicity and clinical
relevance. The issue for clinician and patient is whether
the effect is expected under usual dosing conditions.
A yes means that the usual patient on the usually effective
antidepressant dose of the drug achieves concentrations
of parent drug and/or metabolites that should engage that
specific target to a physiologically/clinically significant
extent given the in vitro affinity of the parent
drug and/or metabolites for that target. If the affinity
for another target is within an order of magnitude of
desired target, then that target is likely also affected
to a physiologically relevant degree. For example, under
usual dosing conditions, amitriptyline achieves concentrations
that engage the norepinephrine uptake pump. At such concentrations,
it also substantially blocks histamine-1 and muscarinic
acetylcholine receptors since it has even more affinity
for those targets than it does for the norepinephrine
uptake pump. Since the binding affinity of amitriptyline
for the 5-HT2A and a-1 norepinephrine receptors and the
serotonin uptake pump are within an order of magnitude
of its affinity for the norepinephrine uptake pump, amitriptyline
at usual therapeutic concentrations for antidepressant
efficacy will also affect those targets. On the other
hand, amitriptyline will not typically affect Na+ fast
channels at usual therapeutic concentrations because there
is more than an order of magnitude (ie, > ten-fold) separation
between its effects on this target versus norepinephrine
uptake inhibition. Nevertheless, an amitriptyline overdose
can result in concentrations which engage this target.
That fact accounts for the narrow therapeutic index of
the tricyclic antidepressants (TCAs) and is the reason
therapeutic drug monitoring to detect unusually slow clearance
is a standard of care when using such drugs.
Most potent effect (ie, effect that occurs at lowest
concentration). See previous footnote for further explanation. |
| The binding affinities
of all drugs listed in the table (except mirtazapine)
are based on the work of Cusack et al and Bolden-Watson
and Richelson. Information on the binding affinities of
mirtazapine (including affinities for 5-HT2A and 5-HT3
receptors) is based on the work of de Boer et al. Although
the publications by the Richelson group did not include
values for the 5-HT2A and 5-HT3 receptors or the other
antidepressants, Elliot Richelson of the Mayo Clinic in
Jacksonville, Florida (personal communication) confirmed
that the other antidepressants would be unlikely to affect
these receptors under usual dosing conditions. |
| Adapted from: Bolden-Watson
C, Richelson E. Life Sci. 1993;52:1023-1029; Cusack
B, et al. Psychopharmacology. 1994;114:559-565;
de Boer T, et al. Neuropharmacology. 1988;27:399-408;
de Boer T, et al. Hum Psychopharmacol. 1995;10:107S-118S;
and Frazer A. J Clin Psychiatry. 1997;58(suppl
6):9-25. |
The following equation explains how dose is
related to the drug concentration, which is the second term
in Equation 2:
(Equation 3)
Concentration = dosing rate (mg/day)/clearance
(mL/min)
In other words, the concentration achieved in
a specific patient is determined by the dosage of the drug
the patient is taking relative to the patient's ability to
clear the drug from the body.
| TABLE
6.3 Sites of Action and Clinical and Physiologic Consequences
of Blockade or Antagonism |
| Site of Action |
Consequence of
Blockade |
| Histamine-1 receptor |
Sedation, antipruritic effect |
| Muscarinic acetylcholine receptor |
Dry mouth, constipation, sinus
tachycardia, memory impairment |
| NE uptake pump |
Antidepressant efficacy, blood
pressure, tremors, diaphoresis |
| 5-HT2A receptor |
Antidepressant efficacy, rapid
eye movement sleep, antianxiety efficacy, anti-extrapyramidal
symptoms |
| a-1 NE receptor |
Orthostatic hypotension, sedation
|
| 5-HT2 uptake pump |
Antidepressant efficacy, nausea,
loose stools, insomnia, anorgasmia |
| a-2 NE receptor |
Antidepressant efficacy, arousal,
libido |
| 5-HT2C receptor |
Antianxiety efficacy, appetite,
¯ motor restlessness |
| 5-HT3 receptor |
Antinauseant |
| Fast Na+ channels |
Delayed repolarization leading
to arrhythmia, seizures, delirium |
| Dopamine uptake pump |
Antidepressant efficacy, euphoria,
abuse potential, antiparkinson activity, aggravation of
psychosis |
| Monoamine oxidase |
Antidepressant activity, decreased
blood pressure* |
| Abbreviations: NE,
norepinephrine; 5-HT, 5-hydroxytryptamine (serotonin);
Na+, sodium. |
| * Hypertensive crisis
(ie, markedly elevated blood pressure) and serotonin syndrome
can occur when monoamine oxidase inhibitors are combined
with noradrenergic and serotonin agonists, respectively. |
| Adapted from: Preskorn
SH. Clinical Pharmacology of Selective Serotonin Reuptake
Inhibitors. Caddo, Okla: Professional Communications,
Inc; 1996;48-49. |
| TABLE 6.4 Comparison
of the Placebo-Subtracted Incidence Rate (%) of Frequent
Adverse Effects for Citalopram, Fluoxetine, Fluvoxamine,
Paroxetine, and Sertraline* |
| Adverse Effect |
Citalopram (n=1063,
n=446)a |
Fluoxetine (n=1730,
n=799)a |
Fluvoxamine (n=222,
n=192)a |
Paroxetine (n=421,
n=421)a |
Sertraline (n=861,
n=853)a |
| Anorexia |
2 |
7.2
|
8.6 |
4.5 |
1.2 |
| Confusionb |
NA |
1.5 |
NA |
1 |
0.8 |
| Constipation |
< placebo |
1.2 |
11.2 |
5.2 |
2.1 |
| Diarrheac |
3 |
5.3 |
-0.4 |
4 |
8.4 |
| Dizzinessd |
< placebo |
5 |
1.3
|
7.8 |
5 |
| Drowsinesse |
8 |
5.9 |
17.2 |
14.3 |
7.5 |
| Dry mouth |
6 |
3.5 |
1.8 |
6 |
7 |
| Dyspepsia |
1 |
2.1 |
3.2 |
0.9 |
3.2 |
| Fatiguef |
2 |
5.6 |
6.2 |
10.3 |
2.5 |
| Flatulence |
NA |
0.5 |
NA |
2.3 |
0.8 |
| Frequent micturition |
NA |
1.6 |
0.6 |
2.4 |
0.8 |
| Headache |
< placebo |
4.8 |
2.9 |
0.3 |
1.3 |
| Increased appetite |
NA |
NA |
NA |
NA |
NA |
| Insomnia |
1 |
6.7 |
4 |
7.1 |
7.6 |
| Nauseag |
8 |
11 |
25.6 |
16.4 |
14.3 |
| Nervousnessh |
3 |
10.3 |
7.6 |
4.9 |
4.4 |
| Palpitationsi |
< placebo |
-0.1 |
NA |
1.5 |
1.9 |
| Paresthesiaj |
NA |
-0.3 |
NA |
2.1 |
1.3 |
| Rashk |
NA |
0.9 |
NA |
1 |
0.6 |
| Respiratoryl |
8 |
5.8 |
-1.3 |
0.8 |
0.8 |
| Sweating |
2 |
4.6 |
-1.3 |
8.8 |
5.5 |
| Tremors |
2 |
5.5 |
6.1 |
6.4 |
8 |
| Urinary retentionm |
< placebo |
NA |
NA |
2.7 |
0.9 |
| Vision disturbances |
< placebo |
1 |
0 |
2.2 |
2.1 |
| Weight gain |
NA |
NA |
NA |
NA |
NA |
| Abbreviations: NA,
not available. |
* Data for fluoxetine,
paroxetine and sertraline is from Preskorn SH. J Clin
Psychiatry. 1995;56(suppl 6):12-21; data for fluvoxamine
is from Compendium of Pharmaceuticals and Specialties.
33rd ed. 1998:922-924; data for citalopram is from Forest
Pharmaceuticals, Inc. prescribing information; 1998. Incidence
of each respective adverse effect for patients taking
each drug minus the incidence for each drugs parallel
placebo control in double-blind, placebo-controlled studies.
The above adverse effect data come from product labeling
as opposed to head-to-head trials. Such data may not necessarily
reflect the actual rate of these adverse effects in clinical
practice or the actual differences between these various
drugs. |
a The
first value is the number of patients on that medication,
while the second represents those treated in the parallel,
placebo group.
bIncludes decreased concentration, memory impairment,
abandoned thinking concentration.
cIncludes gastroenteritis.
dIncludes lightheadedness, postural hypotension,
and hypotension.
eIncludes somnolence, sedation, and drugged
feeling.
fIncludes asthenia, myasthenia, and psychomotor
retardation.
gIncludes vomiting.
hIncludes anxiety, agitation, hostility, akathisia,
and central nervous system stimulation.
iIncludes tachycardia and arrhythmias.
jIncludes sensation disturbances and hypesthesia.
kIncludes pruritus.
lIncludes respiratory disorder, upper respiratory
infection, flu, dyspnea, pharyngitis, sinus congestion,
oropharynx disorder, fever, and chill.
mIncludes micturition disorder, difficulty
with micturition, and urinary hesitancy. |
| TABLE 6.5 Comparison
of the Placebo-Subtracted Incidence Rate (%) of Frequent
Adverse Effects for Bupropion, Imipramine, Mirtazapine,
Nefazodone, and Venlafaxine* |
| Adverse Effect |
Bupropion (n=323,
n=185)a |
Imipramine (n=367,
n=672)a |
Mirtazapine (n=453,
n=361)a |
Nefazodone
(n=393, n=394)a |
Venlafaxine-IR
(n=1033, n=609)a |
Venlafaxine-XR
(n = 357,n = 285)a |
| Anorexia |
-0.1 |
NA |
NA |
NA |
9 |
4 |
| Confusionb |
2.8 |
NA |
2 |
9 |
1 |
2 |
| Constipation |
8.7 |
17.4 |
6 |
6 |
8 |
3 |
| Diarrheac |
-1.8 |
--2.7 |
< placebo |
1 |
1 |
< placebo |
| Dizzinessd |
6.8 |
22.7 |
4 |
23 |
12 |
11 |
| Drowsinesse |
0.3 |
12 |
36 |
11 |
14 |
9 |
| Dry mouth |
9.2 |
47.1 |
10 |
12 |
11 |
6 |
| Dyspepsia |
0.9 |
NA |
< placebo |
2 |
1 |
< placebo |
| Fatiguef |
-3.6 |
7.6 |
3 |
7 |
6 |
1 |
| Flatulence |
NA |
NA |
< placebo |
NA |
1 |
1 |
| Frequent micturition |
0.3 |
NA |
1 |
1 |
1 |
NA |
| Headache |
3.5 |
-8.7 |
< placebo |
3 |
1 |
< placebo |
| Increased appetite |
NA |
NA |
15 |
NA |
1 |
2 |
| Insomnia |
5.3 |
0.4 |
< placebo |
2 |
8 |
6 |
| Nauseag |
4 |
1.3 |
< placebo |
11 |
26 |
21 |
| Nervousnessh |
13.9 |
3.6 |
< placebo |
NA |
12 |
7 |
| Palpitationsi |
4.7 |
NA |
< placebo |
NA |
2 |
< placebo |
| Paresthesiaj |
0.8 |
NA |
NA |
2 |
1 |
NA |
| Rashk |
3.7 |
NA |
NA |
2 |
1 |
NA |
| Respiratoryl |
-2.5 |
-2.3 |
3 |
9 |
NA |
1 |
| Sweating |
7.7 |
11.2 |
< placebo |
NA |
9 |
11 |
| Tremors |
13.5 |
10 |
1 |
1 |
4 |
3 |
| Urinary retentionm |
-0.3 |
4 |
NA |
1 |
2 |
NA |
| Vision disturbances |
4.3 |
5.4 |
< placebo |
12 |
4 |
4 |
| Weight gain |
NA |
NA |
10 |
NA |
NA |
NA |
| Abbreviations: IR,
immediate release; XR, extended release; NA, not available. |
* Data from Preskorn
SH. J Clin Psychiatry. 1995;56(suppl 6):12-21;
Remeron (mirtazapine). Physicians Desk Reference;
1999:2147-2149; and Effexor (venlafaxine hydrochloride).
Physicians Desk Reference; 1999:3298-3302.
The above adverse effect data come from product labeling
as opposed to head-to-head trials. Such data may not necessarily
reflect the actual rate of these adverse effects in clinical
practice or the actual differences between these various
drugs. |
a The
first value is the number of patients on that medication,
while the second represents those treated in the parallel,
placebo group.
bIncludes decreased concentration, memory impairment,
abandoned thinking concentration.
cIncludes gastroenteritis.
dIncludes lightheadedness, postural hypotension,
and hypotension.
eIncludes somnolence, sedation, and drugged
feeling.
fIncludes asthenia, myasthenia, and psychomotor
retardation.
gIncludes vomiting.
hIncludes anxiety, agitation, hostility, akathisia,
and central nervous system stimulation.
iIncludes tachycardia and arrhythmias.
jIncludes sensation disturbances and hypesthesia.
kIncludes pruritus.
lIncludes respiratory disorder, upper respiratory
infection, flu, dyspnea, pharyngitis, sinus congestion,
oropharynx disorder, fever, and chill.
mIncludes micturition disorder, difficulty
with micturition, and urinary hesitancy. |
| TABLE 6.6 The
Most Likely Specific Adverse Effects on Specific SSRIs
Above and Beyond the Parallel Placebo Condition (Percentage
on Drug Minus Percentage on Placebo Based on Registration
Studies)* |
| SSRI |
> 7.5% |
> 10% |
> 15% |
> 20% |
> 25% |
> 30% |
> 35% |
> 40% |
> 45% |
| Citalopram |
Drowsiness
Respiratory
Nausea |
|
|
|
|
|
|
|
|
| Fluoxetine |
|
Nausea
Nervousness |
|
|
|
|
|
|
|
| Fluvoxamine |
Anorexia |
Constipation |
Drowsiness |
|
Nausea |
|
|
|
|
| Paroxetine |
Dizziness
Sweating |
Fatigue |
Drowsiness
Nausea |
|
|
|
|
|
|
| Sertraline |
Insomnia
Diarrhea |
Nausea |
|
|
|
|
|
|
|
| Abbreviations: SSRI,
serotonin selective reuptake inhibitors. |
* Best available
data also suggests that all SSRIs can cause sexual dysfunction
(eg, delayed ejaculation, decreased libido, anorgasmia)
in approximately 30% of patients.
This table is based on Table 6.4.
The above adverse effect data come from product labeling
as opposed to head-to-head trials. Such data may not necessarily
reflect the actual rate of these adverse effects in clinical
practice or the actual differences between these various
drugs. |
Pharmacodynamic Principles Central
to Understanding Antidepressant Options
As a general rule, most drugs (including antidepressants)
act as antagonists at their site(s) of action. Table 6.3 shows
the effects produced by blocking a specific neural mechanism.
If a drug were to act as an agonist at a specific site, then
it would produce the opposite effect to that shown in Table
6.3.
Antidepressants which block neuronal uptake
pumps for serotonin, norepinephrine and dopamine act as indirect
agonists by increasing the concentration of these neurotransmitters
at their respective receptors. Thus, the clinical effects
of uptake inhibitors will be the opposite of those shown in
Table 6.3. For example, by increasing the availability of
serotonin or 5-hydroxytryptamine (5-HT) receptors, serotonin
reuptake inhibitors (SRIs) act as indirect agonists at the
following receptors:
As outlined in Table 6.2, SRIs include all of the serotonin
selective reuptake inhibitors (SSRIs) and the serotonin and
norepinephrine reuptake inhibitor (SNRI), venlafaxine.
The above helps to explain why all SRIs can:
- Decrease rapid eye movement (REM) sleep and shift sleep
architecture from restorative, deep stage IV sleep to light
stage I sleep due to indirect stimulation of the 5-HT2A
receptor9,159,217,226,230
- Decrease appetite and cause motor restlessness due to
indirect stimulation of the 5-HT2C receptor238
- Cause nausea by indirect stimulation of the 5-HT3 receptor.83
The indirect stimulation of one of these 5-HT receptors (or
perhaps another) likely mediates the sexual dysfunction seen
with all SSRIs, including:
- Anorgasmia
- Delayed ejaculation
- Decrease libido.145,147
| TABLE 6.7 The
Most Likely Specific Adverse Effects on Specific Antidepressants
Above and Beyond the Parallel Placebo Condition (Percentage
on Drug Minus Percentage on Placebo Based on Registration
Studies)* |
| Antidepressant |
> 7.5% |
> 10% |
> 15% |
> 20% |
> 25% |
> 30% |
> 35% |
> 40% |
> 45% |
| Bupropion |
Dry mouth
Constipation
Sweating
|
Tremors
Nervousness
|
|
|
|
|
|
|
|
| Imipramine |
Fatigue |
Tremors
Sweating
|
Constipation |
Dizziness |
|
|
|
|
Dry mouth |
| Mirtazapine |
|
Weight gain
Dry mouth
|
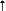 Appetite
Appetite |
|
|
|
Drowsiness |
|
|
| Nefazodone |
Confusion
Respiratory
|
Drowsiness
Vision disturbance
Nausea
Dry mouth
|
|
Dizziness |
|
|
|
|
|
| Venlafaxine-IR |
Insomnia
Anorexia
Constipation
Sweating
|
Nervousness
Dizziness
Dry mouth
|
Drowsiness |
|
Nausea |
|
|
|
|
| Venlafaxine-XR |
Nervousness
Drowsiness
|
Sweating
Dizziness
|
|
Nausea |
|
|
|
|
|
| Abbreviations: IR,
immediate release; XR, extended release. |
* This table is
based on Table 6.5.
The above adverse effect data come from product labeling
as opposed to head-to-head trials. Such data may not necessarily
reflect the actual rate of these adverse effects in clinical
practice or the actual differences between these various
drugs. |
The reverse logic explains the effects
of other antidepressants. For example, nefazodone and mirtazapine
can increase stage IV sleep most likely by blocking the 5-HT2A
receptor.214 Mirtazapine
can also increase appetite and decrease motor restlessness
by blocking the 5-HT2C receptor and can treat nausea by blocking
the 5-HT3 receptor.83
Trazodone is also a 5-HT2A blocker which is consistent with
its widespread use as an antidote for the sleep disturbances
that occur in some patients on SRIs.155
In addition, the effects of mirtazapine and trazodone on sleep
are further amplified by their shared ability to block central
histamine receptors.31,54,58,59,77
The fact that nefazodone and mirtazapine cause minimal, if
any, sexual dysfunction is consistent with their minimal inhibition
of serotonin uptake perhaps coupled with their blockade of
the 5-HT2A receptor.31,54,58,59,77
These issues are further discussed in Chapters 8 and 11.
Some antidepressants, like SSRIs, directly affect only one
site of action at the usual concentration achieved under therapeutic
dosing conditions; other antidepressants affect more than
one site. As shown in Table 6.1, venlafaxine and nefazodone
affect more than one site; the same is also true for bupropion.
Tertiary amine tricyclic antidepressants (TATCAs), such as
amitriptyline, affect six different targets at usual therapeutic
concentrations.
Drugs which affect more than one site of action often act
sequentially (ie, as their concentration increases, they affect
additional targets) which explains why these drugs exhibit
different effects to different degrees at different doses.
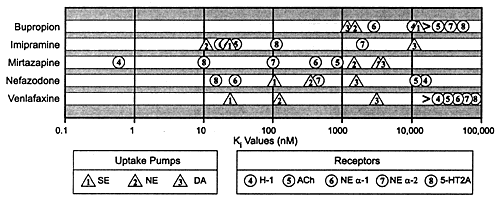
Figure 6.1 illustrates this pharmacologic principle: amitriptyline
can affect multiple sites of action, in contrast to desipramine
and sertraline which are selective for a single (although
different) neural site of action. Parenthetically, some SSRIs
(eg, fluoxetine) affect cytochrome P450 (CYP) enzymes, which
are non-neural sites of action with important pharmacokinetic
drug-drug interaction consequences (Figure 6.2). This issue
is discussed in greater
detail in Chapters 8 and 10.
| FIGURE 6.1 Relative Potency
for Different Sites of Action for Three Different Types
of Antidepressants: Amitriptyline, Desipramine, and Sertraline |
 |
| Abbreviations: NE,
norepinephrine; SE, serotonin; DA, dopamine; H-1, histamine;
ACh, acetylcholine; 5-HT, 5-hydroxytryptamine (serotonin). |
| Based on data from:
Bolden-Watson C, Richelson E. Life Sci. 1993;52:1023-1029
and Cusack B, et al. Psychopharmacology. 1994;114:559-565. |
| FIGURE 6.2 In Vivo Profile
of SSRIs on SE Uptake Inhibition Versus CYP Enzyme Inhibition |
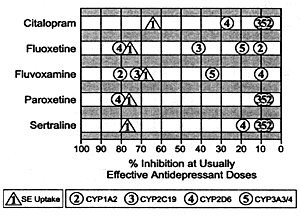
|
| Abbreviations: SSRI,
serotonin selective reuptake inhibitor; SE, serotonin;
CYP, cytochrome P450 enzyme. |
| Based on data from:
Shad MU, Preskorn SH. In: Levy R, et al, eds. Philadelphia,
Pa: Lippincott, Williams &Wilkins. In press. |
The multiple actions of the TATCAs (eg,
amitriptyline, doxepin, imipramine, trimipramine), like the
effects of some newer antidepressants on CYP enzymes, are
now generally considered to be a clinical disadvantage.170,171
To understand the pharmacology of amitriptyline, imagine the
number of single mechanism of action drugs the patient would
have to take to achieve all the effects that can be obtained
with amitriptyline alone (Table 6.8). At low doses (or concentrations),
amitriptyline blocks histamine receptors; at higher doses,
amitriptyline sequentially blocks the other sites as illustrated
in Figure 6.1 and enumerated in Tables 6.2 and 6.3.
| TABLE
6.8 Amitriptyline: Polydrug Therapy in a Single Pill |
| Drug |
Action |
| Chlorpheniramine |
Histamine-1 receptor blockade |
| Cimetidine |
Histamine-2 receptor blockade |
| Benztropine |
Acetylcholine receptor blockade |
| Desipramine |
NE uptake inhibition |
| Nefazodone |
5-HT2A receptor blockade |
| Sertraline |
Serotonin uptake inhibition |
| Prazosin |
a-1 NE receptor blockade |
| Yohimbine |
a-2 NE receptor blockade |
| Quinidine |
Direct membrane stabilization |
| Abbreviations: NE,
norepinephrine; 5-HT, 5-hydroxytryptamine (serotonin). |
The pharmacology of TATCAs such as amitriptyline
served as the blueprint for what effects newer antidepressants
should and should not have and, thus, amitriptyline also served
as the basis for the classification system presented in Table
6.1.181,236
Table 6.2 lists specific neural mechanisms in the order in
which they are affected by amitriptyline; the most potent
mechanism (histamine receptor blockade) being listed at the
top and the least potent (inhibition of sodium [Na+] fast
channels) listed near the bottom. The latter action occurs
at drug concentrations approximately 10 times higher than
those needed to block neural uptake pumps for norepinephrine
and serotonin. While inhibition of neural uptake pumps for
these chemical transmitters is believed to be responsible
for the antidepressant efficacy of the TATCAs, the inhibition
of Na+ fast channels results in slowing of intracardiac conduction
and hence can cause fatal arrhythmias. The potency of TATCAs
for this site of action explains why only modest overdoses
can be lethal in suicide attempts.194
One goal of rational drug development
with regard to newer antidepressants was to widen the therapeutic
index by avoiding the inhibition of Na+ fast channels.170
Another goal was to avoid the adverse effects of TATCAs, which
can be troublesome even if not seriously toxic. For example,
all of the SSRIs share with TATCAs the ability to block the
neural uptake of serotonin, but do not share the ability to
block histamine, muscarinic cholinergic and alpha-1-adrenergic
receptors. For this reason, SSRIs avoid the adverse effects
of sedation, constipation and orthostatic hypotension which
plague the users of TATCAs (Figure 6.3). Other newer antidepressants
engage other neural targets at concentrations achieved under
clinically relevant dosing conditions (Figure 6.4 and Table
6.2).
Pharmacodynamic Drug-Drug Interactions
The clinician can also use Tables 6.2 and
6.3 to anticipate what type of pharmacodynamically mediated
drug interactions are likely to occur when a specific type
of antidepressant is used in combination with other agents.
For example, all SSRIs can interact with MAOIs to cause the
serotonin syndrome.51,87,144,158,228,233
Similarly, all antidepressants that centrally block the histamine
receptor potentiate the adverse cognitive-motor effects of
alcohol.140,151,192
All antidepressants that block the alpha-1-adrenergic receptor
have the potential to aggravate the orthostatic hypotension
caused by other antihypertensive medications.103,192
| FIGURE 6.3 Relative Potency
for Different Sites of Action for the Various Members
of the SSRI Class of Antidepressants |
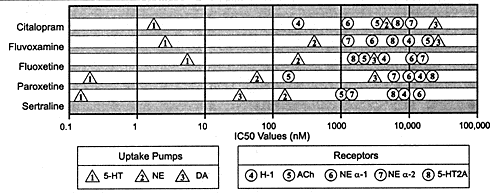 |
| Abbreviations: SSRI,
serotonin selective reuptake inhibitor; 5-HT, 5-hydroxytryptamine
(serotonin); NE, norepinephrine; DA, dopamine; H-1, histamine;
ACh, acetylcholine. |
| Based on data from:
Hyttel J. Nord J Psychiatry. 1993;47(suppl 30)5-12. |
| FIGURE 6.4 Relative Potency
for Different Sites of Action for Non-SSRI Antidepressants:
Bupropion, Imipramine, Mirtazapine, Nefazodone, and Venlafaxine |
 |
| Abbreviations: SE,
serotonin; NE, norepinephrine; DA, dopamine; H-1, histamine;
ACh, acetylcholine; 5-HT, 5-hydroxytryptamine (serotonin). |
| Based on data from:
Bolden-Watson C, Richelson E. Life Sci. 1993;52:1023-1029;
Cusack B, et al. Psychopharmacology. 1994;114:559-565;
de Boer T, et al. Neuropharmacology. 1988;27:399-408;
and de Boer T, et al. Hum Psychopharmacol. 1995;10:107S-118S. |
A Non-neural Target: Cytochrome
P450 Enzymes
As mentioned earlier, CYP enzymes can be a site of action
for a drug.93,94
Although CYP enzymes may be unintended and unnecessary targets
for an antidepressant, they are clinically important because
inhibition of these enzymes (Figure 6.2) carries with it the
liability for causing specific types of pharmacokinetic drug-drug
interactions.
While newer antidepressants were designed to selectively
affect specific neural targets (eg, a specific intake pump)
and avoid others (eg, muscarinic cholinergic receptors), they
were not rationally designed to avoid the inhibition of CYP
enzymes. The reason is simple: the newer antidepressants were
developed before the isolation of the first CYP enzyme in
1988.84 In fact,
the newer antidepressants were synthesized and screened against
targets known in the mid- to late 1970s. Since it is not possible
to avoid "hitting" what is not known, some of the
new antidepressants have subsequently been found to unintentionally
inhibit one or more drug-metabolizing CYP enzyme to a clinically
meaningful degree (Figure 6.2 and Table 6.6).
The five major CYP enzymes mediating oxidative drug metabolism
(in order of importance) are:
- CYP 3A3/4
- CYP 2D6
- CYP 1A2
- CYP 2C9/10
- CYP 2C19.93,94,223
Of these, CYP 3A3/4 and CYP 2D6 are responsible for approximately
50% and 30% of known oxidative drug metabolism, respectively.115,255
Since the isolation of the first CYP enzyme,
knowledge in this area has exploded. Drugs such as terfenadine
(Seldane) and mibefradil (Posicor) have been withdrawn from
the US market as a result of CYP enzyme-mediated drug interactions.
Testing for effects on CYP enzymes is now part of rational
drug development and is done to avoid developing new agents
that inadvertently inhibit these enzymes.170
Similar testing is also being done on currently available
drugs to determine which CYP enzymes are important for their
clearance and whether they induce or inhibit specific CYP
enzymes.
Such testing is generally done in two steps:
- First, an in vitro approach is used to determine
the ability of a CYP enzyme to metabolize a drug (Table
6.9) and conversely to estimate the potential (ie, potency)
for a new candidate drug to inhibit a CYP enzyme
- Second, formal drug-drug interaction studies in humans
are performed to establish whether clinically meaningful
inhibition occurs under usual dosing conditions (Tables
6.10 and 6.11).93,94,223
This technology thus yields two sets of complementary data
that can be used to predict clinically meaningful CYP enzyme-mediated
drug interactions, as illustrated in Figure 6.5:
- The first set of data determine whether a drug inhibits
a specific CYP enzyme and to what extent under clinically
relevant dosing conditions.
- The second set of data establish which drugs are dependent
on which CYP enzyme for oxidative metabolism.
Table 6.10 summarizes the effects of newer antidepressants
on these five CYP enzymes. Table 6.9 lists the drugs known
to be metabolized by these CYP enzymes.
The principal distinguishing characteristic
among the various SSRIs is their differential effects on CYP
enzymes.93,94,170,223
While all five drugs block the neural uptake of serotonin
and avoid effects on other neural targets, they differ substantially
with regard to their effects on CYP enzymes. Fluoxetine, fluvoxamine
and paroxetine all inhibit one or more CYP enzymes to a clinically
significant degree at their usually effective antidepressant
dose, whereas citalopram and sertraline do not. Since the
inhibition of CYP enzymes conveys no clinical benefit to the
best of our knowledge, the absence of substantial effects
on CYP enzymes conveys a distinct advantage for citalopram
and sertraline relative to other SSRIs.
Pharmacodynamic Principles Central
to Understanding Antidepressant Options
In general, all antidepressants are similar with regard to
a number of pharmacokinetic parameters (ie, absorption and
distribution). The clinically important pharmacokinetic differences
among these drugs include:
- Which CYP enzymes mediate their metabolism? This information
is listed in Table 6.9 and can be used to predict CYP enzyme-mediated
drug-drug interactions in which the antidepressant is the
target rather than the cause of the interaction.
- Some antidepressants (eg, nefazodone) are converted into
metabolites with clinically important effects.103,139
- Difference in half-lives of the various antidepressants.103,176
This issue is arguably the single most important pharmacokinetic
difference among antidepressants beyond their effect on
CYP enzymes.
| TABLE
6.9 Effect of Cytochrome P450 Enzymes on Specific Drugs
(ie, Metabolism) |
| CYP 1A2 |
|
| Antidepressants |
Amitriptyline,
clomipramine, imipramine |
| Antipsychotics |
Clozapine,*
olanzapine,* thioridazine* |
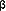 -Blockers -Blockers |
Propanolol |
| Opiates |
Methadone* |
| Miscellaneous |
Caffeine,*
paracetamol, tacrine,* theophylline,* R-warfarin* |
| CYP 2C9/10 |
|
| Miscellaneous |
Phenytoin,* S-warfarin,* tolbutamide* |
| CYP 2C19 |
|
| Antidepressants |
Citalopram,* clomipramine, imipramine |
| Barbiturates |
Hexobarbital, mephobarbital,
S-mephenytoin* |
| -Blockers |
Propranolol |
| Benzodiazepines |
Diazepam |
| CYP 2D6 |
|
| Antiarrhythmics |
Encainide,* flecainide,* mexiletine,
propafenone |
| Antipsychotics |
Haloperidol (minor), molindone,
perphenazine,* risperidone,* thioridazine (minor) |
| -Blockers |
Alprenolol, bufuralol, metoprolol,*
propranolol, timolol |
| Miscellaneous |
Debrisoquine,* 4-hydroxyamphetamine,
perhexiline,* phenformin, sparteine* |
| Opiates |
Codeine,* dextromethorphan,*
ethylmorphine |
| SSRIs |
Fluoxetine, N-desmethylcitalopram,
paroxetine* |
| TCAs |
Amitriptyline,* clomipramine,*
desipramine,* imipramine,* N-desmethylclomipramine |
| Other antidepressants |
Venlafaxine,* mCPP metabolite
of nefazodone* and trazodone* |
| CYP 3A3/4 |
|
| Analgesics |
Acetaminophen, alfentanil |
| Antiarrhythmics |
Amiodarone, disopyramide, lidocaine,
propafenone, quinidine |
| Anticonvulsants |
Carbamazepine,* ethosuximide |
| Antidepressants |
Amitriptyline, clomipramine,
imipramine, nefazodone,* sertraline,* O-desmethylvenlafaxine* |
| CYP 3A3/4 |
|
| Antiestrogens |
Docetaxel, paclitaxel, tamoxifen* |
| Antihistamines |
Astemizole,* loratadine,* terfenadine* |
| Antipsychotics |
Quetiapine,* clozapine |
| Benzodiazepines |
Alprazolam,* clonazepam, diazepam,
midazolam,* triazolam* |
| Calcium channel
blockers |
Diltiazem,* felodipine,* minodipine,
nicardipine, nifedipine,* niludipine, nisoldipine, nitrendipine,
verapamil* |
| Immunosuppressants |
Cyclosporine,* tacrolimus (FK506-macrolide) |
| Local anesthetics |
Cocaine, lidocaine |
| Macrolide antibiotics |
Clarithromycin, erythromycin,
triacetyloleandomycin |
| Steroids |
Androstenedione, cortisol,* dehydro-3-epiandrosterone,
dexamethasone, estrogen,* testosterone,* estradiol,* ethinylestradiol,
progesterone |
| Miscellaneous |
Benzphetamine, cisapride,* dapsone,*
lovastatin, omeprazole (sulfonation) |
| Abbreviations:
CYP, cytochrome P450 enzyme; SSRI, selective serotonin
reuptake inhibitor; TCA, tricyclic antidepressants; mCPP,
meta-chlorophenylpiperazine. |
| * Principal
CYP enzyme. |
| NOTE:
Such lists are not comprehensive since the CYP enzyme(s)
responsible for biotransformation is known for only approximately
20% of marketed drugs. The reason is that many drugs were
developed before the necessary knowledge and technology
existed. Some drugs are listed under more than one CYP
enzyme. That does not necessarily mean that each of these
enzymes contributes equally to the elimination of the
drug. One enzyme may be principally responsible based
on substrate affinity and capacity and abundance of the
enzyme. |
| Adapted
from: Preskorn SH. Clinical Pharmacology of Selective
Serotonin Reuptake Inhibitors. Caddo, Okla: Professional
Communications, Inc; 1996:158-159. |
| TABLE 6.10 The
Inhibitory Effect of Newer Antidepressants at Their Usually
Effective Minimum Dose on Specific CYP Enzymes |
|
|
No
or Minimal Effect
(< 20%)* |
Mild
(20%-50%)* |
Moderate
(50%-150%)*
|
Substantial
(> 150%)* |
| Bupropion |
? |
?
|
? |
2D6 |
| Citalopram |
1A2,
2C9/10, 2C19, 3A3/4 |
2D6
|
|
|
| Fluoxetine |
1A2 |
3A3/4 |
2C19 |
2D6,
2C9/10 |
| Fluvoxamine |
2D6 |
|
3A3/4 |
1A2,
2C19 |
| Nefazodone |
1A2,
2C9/10, 2C19, 2D6 |
|
|
3A3/4 |
| Paroxetine |
1A2,
2C9/10, 2C19, 3A3/4 |
|
|
2D6 |
| Sertraline |
1A2,
2C9/10, 2C19, 3A3/4 |
2D6 |
|
|
| Venlafaxine |
1A2,
2C9/10, 2C19, 3A3/4 |
2D6 |
|
|
| Mirtazapine based
on in vitro modeling is unlikely to produce clinically
detectable inhibition of these five CYP enzymes. However,
no in vivo studies have been done to confirm that prediction. |
| * Percent increase
in plasma levels of a coadministered drug dependent on
this CYP enzyme for its clearance. The potential in
vivo effects of bupropion on CYP enzymes other than
2D6 have not been studied and thus are unknown. |
| Adapted from: Harvey
A, Preskorn SH. J Clin Psychopharmacol. 1996;16:273-285,
345-355; and Shad MU, Preskorn SH. In: Levy R, et al,
eds. Philadelphia, Pa: Lippincott, Williams & Wilkins;
2000:563-577. |
The Clinical Importance of Half-Life
Half-life is the time needed to decrease plasma drug concentration
by 50% after drug discontinuation. As a general rule, steady-state
is achieved when the drug has been administered at a stable
dose for a period equal to 5 times its half-life. The same
is true for washout following drug discontinuation. Thus,
half-life is an important determinant of how long it will
take to achieve maximal drug effect once started, and how
long the effect will persist once the drug is stopped.175
The optimal dosing schedule for antidepressants is virtually
never established empirically. Instead, the half-life is almost
always used to determine the dosage schedule for a drug in
the clinical trials required for its registration. That is
done to maximize the chance for showing acceptable efficacy
and tolerability. Since the FDA requires that the dosing schedules
in the package inserts be based on the schedules used in the
clinical trials, that means the half-life of the drug determines
the recommended dosing schedule contained in the package insert.
In general, the half-life of most antidepressants is approximately
24 hours. For this reason, most of these drugs were administered
once a day during clinical trials.103,176
For the same reason, steady-state and virtually complete washout
are achieved within 5 days of starting or stopping most of
the newer antidepressants.
A few antidepressants have half-lives shorter than 24 hours
including:
- Bupropion
- Fluvoxamine
- Nefazodone
- Venlafaxine.103
TABLE 6.11 —
Summary of Formal In Vivo Studies of the Effects
of Different SSRIs on CYP 2D6 Model Substrates |
| SSRI |
Author |
N |
SSRI Treatment:
Dose (mg/day) x
Duration (days) |
Substrate |
Substrate
Dosing |
Results |
(AUC2-AUC1)
÷ AUC1 |
DM/DO* |
EMs
to PMs |
| Citalopram |
Gram |
8 |
40 x 10 |
DMI |
Single Dose |
47% |
|
|
| Fluoxetine |
Lam |
8 |
60 x 8 |
DM |
Single dose |
|
3484% |
62.5% |
| Amchin |
12 |
20 x 28 |
DM |
Single dose |
|
1711% |
|
| Bergstrom |
6 |
60 x 8 |
DMI |
Single dose |
640% |
|
|
| 6 |
60 x 8 |
IMI |
Single dose |
IMI 235%
DMI 430% |
|
|
| Preskorn |
9 |
20 x 21 |
DMI |
21 days |
380% |
|
|
| Otton |
19 |
37 ± 17 x 21 |
DM |
Single dose |
|
|
95% |
| Mace |
11 |
20 x 28 |
mCPP |
7 days |
820% (270%) |
|
|
| Fluvoxamine |
Lam |
6 |
100 x 8 |
DM |
Single dose |
|
> 6% |
0% |
| Spina |
6 |
100 x 10 |
DMI |
Single dose |
14% |
|
|
| Paroxetine |
Alderman |
17 |
20 x 9 |
DMI |
9 days |
421% |
|
|
| Brosen |
9 |
20 x 8 |
DMI |
Single dose |
364% |
|
78% |
| Albert |
10 |
30 x 4 |
IMI |
Single dose |
IMI 74%
DMI 327% |
|
|
| Lam |
8 |
20 x 8 |
DM |
Single dose |
|
3943% |
50% |
| Ozdemir |
8 |
20 x 10 |
PRZ |
Single dose |
595% |
|
|
| Sertraline |
Alderman |
17 |
50 x 9 |
DMI |
8 days |
37% |
|
|
| Jann |
4 |
50 x 7 |
DMI |
7 days |
0% |
|
|
| Preskorn |
9 |
50 x 21 |
DMI |
21 days |
23% |
|
|
| Solai |
13 |
50 x > 5 |
NTP |
Chronic dosing |
14% |
|
|
| Ozdemir |
19 |
94 ± 26 x 24 ±
17 |
DM |
Single dose |
0% |
|
0% |
| Sproule |
6 |
108 ± 49 x 21 |
DM |
Single dose |
5% |
22% |
0% |
| Lam |
7 |
100 x 8 |
DM |
Single dose |
|
28% |
0% |
| Kurtz |
6
6 |
150 x 8
150 x 8 |
IMI
DMI |
Single dose
Single dose |
68%
54% |
|
|
| Zussman |
13 |
150 x 29 |
DMI |
Single dose |
70% |
|
|
| Abbreviations: SSRI,
serotonin selective reuptake inhibitor; CYP, cytochrome
P450 enzyme; AUC2, area under the curve of the substrate
with SSRI; AUC1, area under the curve of the substrate
without SSRI; DM, dextromethorphan; DO, dextrorphan; EM,
extensive metabolizer; PM, poor metabolizer; DMI, desipramine;
IMI, imipramine; mCPP, meta-chlorophenylpiperazine (a
metabolite of nefazodone and trazodone); PRZ, perphenazine;
NTP, nortriptyline. |
* Percent increase.
60 mg/day for 8 days is a loading-dose strategy used
to approximate the plasma levels of fluoxetine and norfluoxetine
achieved under steady-state conditions on a dose of 20
mg/day.
820% is based on all the data. If the two highest increases
are excluded, the average was 270%. |
| Adapted from: Preskorn
SH. J Psychopharmacology. 1998;12:S89-S97. |
For this reason, these drugs were tested using either a twice-a-day
or even a three-times-a-day schedule during registration
trials.
| FIGURE 6.5 How
Knowledge of CYP Enzymes Will Simplify Understanding of
Pharmacokinetic Interactions |
| Drug A |
---Affects*---> |
P450 enzyme X |
| P450 enzyme X |
--Metabolizes--> |
Drugs B, C, D, E,
F |
| Therefore, Drug
A |
---Affects*---> |
Drugs B, C, D, E,
F |
|
| Abbreviations: CYP, cytochrome
P450 enzyme. |
| * Could be inhibition or induction. |
| Adapted from: Preskorn SH. Clinical
Pharmacology of Selective Serotonin Reuptake Inhibitors.
Caddo, Okla: Professional Communications, Inc; 1996;156. |
Since frequent dosing schedules are perceived as a disadvantage,
extended-release formulations of bupropion and venlafaxine
have been developed. Although the half-life of the drug remains
the same, the extended absorption of these formulations produce
reasonably stable blood levels of the drug over a more prolonged
dosing interval.
The only antidepressant with a half-life substantially in
excess of 24 hours is fluoxetine. The parent drug has a half-life
of 2 to 4 days and norfluoxetine, its active metabolite, has
a half-life of 7 to 15 days.170
This half-life is even longer at doses above 20 mg/day in
physically healthy older patients.197
These half-lives mean that fluoxetine is essentially an oral
depot drug that requires more than a month to:
- Reach steady-state once started
- Clear once stopped.
The practitioner must realize that once
fluoxetine has achieved steady state, its effects cannot rapidly
be reversed and that there will be an appreciable interval
after its discontinuation during which fluoxetine (or norfluoxetine)
can affect the response of the patient to other drug treatment,
either through blockade of the serotonin uptake pump or through
the inhibition of specific CYP enzymes (Chapter 10).
Antidepressant Withdrawal Syndrome
A general rule in clinical psychopharmacology is that the
brain adapts to the presence of drugs. For example, uptake
inhibitors produce downregulation of the receptors for the
specific neurotransmitter whose uptake pump has been inhibited
(eg, some serotonin receptors in the case of serotonin uptake
inhibitors and beta-adrenergic receptors in the case of norepinephrine
uptake inhibitors).17
In fact, such downregulation has been postulated to mediate
the antidepressant efficacy of these drugs.
Such downregulation likely also mediates the withdrawal syndromes
that can be seen when some of these antidepressants are abruptly
stopped. Anticholinergic withdrawal syndrome can be seen when
high-potency muscarinic cholinergic receptor blockers (ie,
TATCAs) are abruptly stopped. The symptoms of anticholinergic
withdrawal syndromes (sometimes called "cholinergic rebound")
are listed in Table 6.12.
| TABLE 6.12
Symptoms of Anticholinergic Withdrawal Syndrome
|
- Loose stools
- Urinary frequency
- Headache
- Hypersalivation
|
|
|
| TABLE 6.13 Symptoms of
Serotonin Reuptake Inhibitor Withdrawal Syndrome |
Serotonin reuptake inhibitor
withdrawal syndrome can be remembered by using the
mnemonic FLUSH:
- Flu-like:
Fatigue
Myalgia
Loose stools
Nausea
- Lightheadedness/dizziness
- Uneasiness/restlessness
- Sleep and sensory disturbances
- Headache
|
|
The SRI withdrawal syndrome can be seen
when serotonin uptake inhibitors are stopped (Table 6.13).26,
38, 53,
78, 125,
126, 133,
198, 209, 210,
219, 229,
272 The SRI withdrawal syndrome is more clinically
important than is the anticholinergic withdrawal syndrome
because it is more common, more severe, and more easily misdiagnosed.
It can mimic worsening of the underlying depression or even
as the emergence of mania. Such misdiagnosis can lead to inappropriate
treatment.38,125,126,128,209,210
The diagnosis is confirmed when the symptoms remit (typically
within 12 to 24 hours) after restarting the SRI. After that,
a more gradual taper can be done to wean the patient off the
drug.
Risk factors for antidepressant withdrawal syndromes are:
- Time on drug
- Potency of drug
- Half-life of drug.53,198,209
As mentioned above, the shorter the half-life,
the more likely the drug will wash out before the brain has
had an opportunity to re-equilibrate (eg, upregulation of
receptors) and, hence, the more likely that withdrawal symptoms
will occur after drug discontinuation. Thus, the order of
likelihood of the SRI withdrawal syndrome following abrupt
discontinuation of an SRI is: fluvoxamine, paroxetine, and
venlafaxine > citalopram and sertraline, > fluoxetine.53,198,209,219
The SRI withdrawal syndrome rarely occurs after nefazodone
discontinuation.
Paroxetine and nefazodone deserve additional comment since
the risk of an SRI withdrawal syndrome does not appear to
correlate with their half-lives. In the case of paroxetine,
the incidence of the SRI withdrawal syndrome is higher than
might be expected for a drug with a half-life of 24 hours.
However, the half-life of paroxetine is a function of its
plasma levels. At low levels, the half-life of paroxetine
is only 10 hours (ie, shortest of all the SRIs) due to the
fact that it is preferentially metabolized by CYP 2D6 at low
levels.93,94,170
CYP 2D6 is a high-affinity, but low-capacity, enzyme for the
metabolism of paroxetine and is inhibited by paroxetine. For
this reason, this CYP enzyme becomes saturated at therapeutic
levels of paroxetine (ie, autoinhibition), and CYP 3A3/4 becomes
the primary mechanism for its elimination. CYP 3A3/4 is a
low-affinity, but high-capacity, enzyme and hence produces
a paroxetine half-life of 24 hours. When paroxetine is discontinued,
its clearance accelerates as its level falls, and that likely
accounts for the increased likelihood of an SRI withdrawal
syndrome following its discontinuation.
In the case of nefazodone, the incidence of an SRI withdrawal
syndrome is less than might be expected given its short half-life
of 4 hours. There are several possible explanations that are
not mutually exclusive:
- Nefazodone has several active metabolites which may cushion
the withdrawal state, particularly the triazoledione metabolite,
which has a considerably longer half-life than the parent
drug, nefazodone.103
- Nefazodone is relatively weak as an SRI. Its most potent
action is 5-HT2A blockade, which does not appear to cause
a withdrawal liability. In fact, pharmacologically
relevant serotonin uptake inhibition requires a daily dose
of nefazodone of at least 500 mg/day and possibly more.150
Either of these reasons may account for why patients treated
with nefazodone at doses below 500 mg/day have a minimal risk
of experiencing the SRI withdrawal syndrome following its
discontinuation.
|



