| Preskorn.com | Printed from: http://www.preskorn.com/books/ssri_s3.html |
| Clinical Pharmacology of SSRI's 3 - Basic Neuropharmacology of SSRIs |
|
|
|
|
Potency and selectivity are fundamental pharmacological concepts essential to understanding the basic neuropharmacology and the clinical psychopharmacology of serotonin selective reuptake inhibitors (SSRIs) including:
In this section, we will:
How In Vitro Studies Are Done to Determine Potency
As discussed in Section 2, the goal in developing the SSRIs is to design drugs capable of inhibiting the neuronal uptake pump for serotonin as with the TCAs, but at the same time, avoiding actions on other neural mechanisms. The first step involves isolating these neural SOAs so that the effects of drugs on them can be studied in vitro.34,129 One approach is to isolate the neuronal uptake pump for serotonin by homogenizing brain regions rich in serotonin terminal fields. The homogenization process lysises the neuronal membranes in such a way that the membrane can close back on itself to form synaptosome preparations which retain the functional integrity of the serotonin uptake pump. The pumps allow the synaptosomes to concentrate serotonin by taking it up from the fluid in which the synaptosomes are suspended. By radioactively tagging the serotonin, the rate of uptake can measure by adding tagged serotonin to the suspension for a specified period of time, then centrifuging and counting radioactivity in the synaptosomal pellet and expressing the result as the amount of radioactivity per milligram of protein. The ability of different drugs to slow or inhibit the pump can then be studied by adding different concentrations of a specific drug to identical aliquots of the same synaptosomal preparation and studying ability of the synaptosomal preparation to take up radioactive serotonin as a function of the concentration of the inhibitor which has been added. All other variables, besides the amount of inhibitor added, are kept the same among the different aliquots.
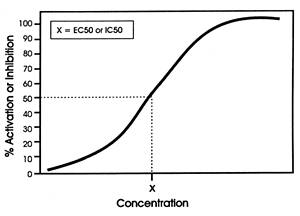
The results from such a study are plotted in Figure 3.1 as a classic concentration-response curve in which the Y-axis is the response (ie, effect of the drug) and the X-axis is increasing concentration of the investigational drug (ie, the potential inhibitor). In the case of the serotonin uptake pump, the effect is the degree of slowing or inhibition of the uptake of the radioactive serotonin into the synaptosomes. This approach represents a biological assay of the effect of the drug on its SOA rather than simply the binding affinity of the drug for the SOA.
| FIGURE 3.1 Generic Curve of a Drugs Concentration-dependent Effect on Specific SOA* |
| |
| * The effect (ie, activation or inhibition) of the drug on the site of action (SOA) is the drugs mechanism of action (MOA). |
In another version of Figure 3.1, the Y-axis can be the binding affinity of the drug for a specific SOA (eg, the histamine1 receptor) rather than its effect on the site.66 In this case, the affinity of the drug for a receptor is measured by its ability to displace a radioactive tagged ligand. From a binding assay, one cannot tell whether the drug is an agonist or an antagonist at that specific SOA; instead, only the affinity of the drug for the receptor is determined.
In either approach, the inflection point is a reproducible measure of the drug's affinity for the site or its effect on the site and hence can be used for comparison purposes across different drugs (ie, relative in vitro potency for that SOA).
As discussed in Section 2, such studies have been done as part of the development process of all of the SSRIs to determine what chemical structure will:
|
Results of In Vitro Studies Done on the Effects of Different SSRIs on Different Biogenic Amine Uptake Pumps
Tables 3.1, 3.2 and 3.3 show the results from three different in vitro studies comparing the effects of four representative TCAs and all five SSRIs on the neuronal uptake pumps for serotonin, norepinephrine and dopamine. As can be seen, the SSRIs are all more potent inhibitors of serotonin uptake than are the TCAs, with the exception of clomipramine, which is less potent than paroxetine or sertraline, approximately equal to citalopram, and more potent than fluoxetine or fluvoxamine.
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The results from the three studies also illustrate the variability that can be obtained in terms of rank order, particularly when the drugs are relatively close in potency. In Study 1, sertraline is approximately twice as potent as paroxetine, whereas in the other two studies, paroxetine is 2- to 5-times more potent than sertraline. Therefore, the rank order shown on the bottom half of the table can change somewhat from one study to the next.
Nonetheless, SSRIs are all substantially more potent in terms of their affinity for the serotonin pump compared with their affinity for or action on any other neurotransmitter pumps or neuroreceptors. When drugs are this selective, differences in potency after a point become clinically irrelevant since the concentration can be adjusted to achieve inhibition of the desired target without affecting any other target. This fact is the essence of the concept of pharmacological selectivity (Figure 3.2).
In Table 3.2, the results for the same drugs from the same studies are shown with regard to their inhibition of the norepinephrine reuptake pump. As can readily be seen, the TCAs are substantially more potent with regard to this action in comparison to all of the SSRIs. As shown on the bottom half, all the SSRIs are two to three orders of magnitude less potent than is the TCA, desipramine, in terms of the ability to inhibit the norepinephrine uptake pump.
In Table 3.3, results are shown for
the inhibition of the dopamine uptake pump. None of the TCAs
or the SSRIs have substantial action on this neurotransmitter
pump. Although sertraline is consistently the most potent,
it is still 100 times less potent in terms of inhibiting the
dopamine versus the serotonin uptake pump. That means the
physician would have to increase the dose (ie, the concentration)
of sertraline 100 times higher than that needed to inhibit
the serotonin uptake pump before a comparable effect would
be achieved on the dopamine uptake pump. Thus, the ratios
shown in the bottom of Table 3.3 can
be misleading if not viewed within the context of the actual
affinity of the drug for a secondary SOA relative to its affinity
for its primary SOA and relative to the clinically relevant
concentration needed to produce the desired clinical effect.
Recall that citalopram and fluoxetine are marketed as racemates
(see Section 2). The values shown
in the above tables for uptake inhibition are
for the racemates of these two SSRIs. Table
3.4 shows the value for the individual enantiomers of
each of these SSRIs and their major metabolite.
| FIGURE 3.2 Selectivity Ratios for a Series of Uptake Inhibitors Measured In Vitro |
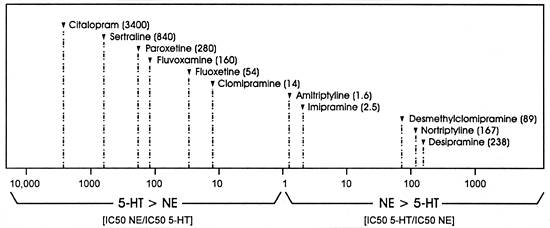 |
| Data from reference: 129 |
The Concept of Selectivity as Related to Effects on Different Biogenic Amine Uptake Pumps
The concept of selectivity is further illustrated in Table 3.5. In this table, the affinity of a specific drug for the norepinephrine uptake pump is divided by its affinity for the serotonin uptake pump. As seen in Tables 3.1, 3.2 and 3.3, the more potent a drug, the smaller the concentration needed to affect or bind to an SOA. Thus, the less potent effect is a larger number (ie, more concentration is needed to produce the same degree of effect), and the more potent effect is the smaller number. In Table 3.5, the ratio is appreciably greater than "1" for all SSRIs, whereas the ratio for all TCAs, except clomipramine, is considerably smaller than "1." SSRIs are considerably more potent at inhibiting the serotonin uptake pump than the norepinephrine uptake pump, whereas the opposite is true for the TCAs, with the exception of clomipramine.
The more the ratio diverges from "1" in either direction, the more selective the drug is for one pump over the other. For example, all SSRIs, with the exception of fluoxetine, are more than 100 times more potent at inhibiting the serotonin versus the norepinephrine uptake pump, whereas the converse is true for the TCA, desipramine. A concentration of any SSRI that will produce substantial inhibition of the serotonin uptake pump will produce no physiologically meaningful inhibition of the norepinephrine uptake pump. The converse will be true for TCAs such as desipramine. Clinically, such selectivity ratios translate into being able to produce all the physiological effects mediated by inhibiting one pump without causing any effects that will be produced by inhibiting the other uptake pump.
Figure 3.2 graphically illustrates the same point. In this figure, a value of "1" means that the drug will inhibit both uptake pumps at the same concentration (ie, no selectivity with regard to effect on these two SOAs). For illustration purposes, the ratio of the right side of the figure is the potency for inhibiting the uptake of serotonin divided by the potency for inhibiting the uptake for norepinephrine, while the inverse is demonstrated on the left side of the figure. This approach is taken so that the ratios will become larger in either direction and hence may be intuitively simpler to understand. In this figure, desipramine is 238 times more potent at inhibiting the norepinephrine uptake pump versus the serotonin uptake pump, whereas all the SSRIs, with the exception of fluoxetine, are over 100 times more potent at inhibiting the serotonin versus the norepinephrine uptake pump.
Affecting any SOA can cause adverse as well as beneficial effects. The physiological responses mediated by activation or inhibition of these and other SOAs are listed in Table 3.6. The goal of rational drug development is to be able to produce drugs that affect the SOA necessary to mediate the desired effect without affecting an SOA that is not critical to producing the desired effect. Affecting unnecessary SOAs can lead to unnecessary adverse effects and an increased potential for causing pharmacodynamic drug-drug interactions.
Potency Relates to Concentration, Not Dose
There is a frequent misconception that potency refers to the dose of a drug needed to produce an effect. That is wrong. Instead, it refers to the concentration of a drug needed to produce an effect. Two drugs may be able to produce exactly the same effect, but the concentration needed of each drug may be quite different. The drug that requires a lower concentration to achieve the same magnitude of effect is the more potent drug regardless of the dose needed to achieve that concentration.
Although dose is sometimes used as the reference point, it is usually because the concentration has not been measured or the author may not be aware of how misleading a dose comparison can be. The concentration achieved by a given dose of a drug is dependent on the bioavailability and elimination rate of the drug. A drug that has lower bioavailability and/or a faster clearance will require a higher dose to produce the same concentration as a drug which has a greater bioavailability or a slower clearance. The critical issue for the SOA is not what dose is taken, but what concentration is achieved at the SOA.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 3.7 illustrates how misleading dose can be, using SSRIs as examples of this basic pharmacological principle. This table shows:
As can be seen, there is little correlation between the dose of the drug and the plasma concentration achieved. For example, the combined plasma concentration of fluoxetine and its active metabolite, norfluoxetine, is 10 times higher than the concentration of sertraline even though the dose of fluoxetine is 2.5 times less than the dose of sertraline. If the comparison was made on the basis of dose, fluoxetine were erroneously appear to be more potent than sertraline as an inhibitor of serotonin uptake. While the dose of SSRIs does not correlate with their in vitro potency, there is a clear correlation between the in vitro potency of the drug and the plasma level of each drug needed to produce relatively comparable serotonin uptake inhibition (ie, lower plasma concentrations of the more potent SSRIs [eg, paroxetine, sertraline] are needed in comparison to higher concentrations of the less potent SSRIs [eg, citalopram, fluoxetine, fluvoxamine]).
The results in Table 3.7 are of clinical and research interest. Each SSRI, at the dose found to be its usually effective, minimum dose based on double-blind, placebo-controlled studies, produces approximately 70% to 80% inhibition of the serotonin uptake pump using the platelet as a surrogate marker. This finding is consistent with the concept that the inhibition of this pump is relevant to the antidepressant efficacy of these drugs and suggests that approximately 70% to 80% inhibition of this pump is usually necessary to produce an antidepressant effect. Higher doses of these drugs do not produce a greater antidepressant response on average (ie, a flat dose-response curve for antidepressant efficacy), but do increase the incidence and severity of adverse effects mediated by excessive serotonin uptake inhibition (eg, agitation, loose stools, nausea). (For more details on this issue, refer to Figures 5.1 and 5.2 later in this book.) These two observations, coupled with the results shown in Table 3.7, indicate that inhibition of the serotonin uptake pump by substantially more than 80% produces a greater increase in adverse effects than an increase in antidepressant efficacy and is one reason to avoid the temptation to use a dose higher than the usually effective dose before it has been given an adequate trial (ie, approximately 4 weeks).
Obviously, the results in Table 3.7 pertain to the average patient. A patient who has a rapid clearance of the drug may need a higher than average dose to achieve an effective concentration, whereas a patient who has a slow clearance may do better in terms of the ratio of efficacy-to-adverse effects on a dose lower than usually effective, minimum dose. (For more details on this issue, refer to the Therapeutic Drug Monitoring discussion in Section 5.)
The Concept of Selectivity as Related to Effects on Different Neuroreceptors
The goal of development of SSRIs is not only to avoid affecting the norepinephrine and dopamine uptake pumps, but a variety of neuroreceptors (in contrast to the TCAs). Figure 3.3 illustrates how well that goal is accomplished using clomipramine as the reference TCA. Shown in this figure are the binding affinities of 7 different chemical agents (ie, clomipramine and its primary metabolite, desmethylclomipramine, and all SSRIs) for 5 clinically important neuroreceptors as well as the 3 biogenic amine uptake pumps. The X-axis is nanomolar concentration on an algorithmic scale so that each vertical line represents an increase in concentration of ten times the previous one. The further the distance between the drug's affinity for one SOA and the next, the greater its selectivity for affecting that target without affecting the next potential target.
| FIGURE 3.3 In Vitro Profile of Antidepressants |
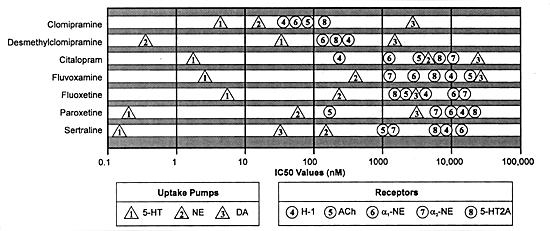 |
| Adapted from reference: 129 |
Clomipramine differs from the other tertiary amine TCAs (eg, amitriptyline, doxepin, imipramine) in that its most potent action is on a site believed to mediate efficacy in major depression and also obsessive-compulsive disorders (ie, the serotonin uptake pump). In contrast, the other tertiary amine TCAs block the histamine receptor as their most potent action, which is why their most potent effect is sedation and why they can potentiate the effect of other sedative agents, including alcohol.76,123,155,188,211,251 Clomipramine, like the other tertiary amine TCAs, has little separation between potency for effects on multiple SOAs such as the norepinephrine uptake pump, the histamine-1, a-1 adrenergic, acetylcholine, 5-HT2A neuroreceptors and the dopamine uptake pump (Figure 3.3). The widest gap for clomipramine is approximately 300 times for the inhibition of the serotonin versus the dopamine uptake pump. That difference is such that clomipramine is unlikely to produce meaningful effects on the dopamine uptake pump at doses which substantially inhibit the serotonin uptake pump. In contrast, the difference between its affinity for the serotonin uptake pump and the other neural SOAs (eg, the norepinephrine uptake pump and various neuroreceptors) is 10-fold or less. That difference is small enough that effects on these sites may occur under clinically relevant conditions and thus can contribute to the clinical pharmacology of the drug. If clinical effects mediated by the drug's action on these sites are unnecessary for the desired clinical effects, these effects will be termed "side-effects" and may range from being a nuisance to treatment-limiting problems to serious adverse effects.
Although not shown in Figure 3.3, clomipramine, like the other TCAs, is also capable of inhibiting fast sodium channels.57 The potency of the drug for this action is such that it occurs to a clinically meaningful extent in the healthy individual only at concentrations above its therapeutic range for antidepressant efficacy and the probable reason for its dose- and, hence, concentration-dependent seizure risk and cardiac arrhythmia risk.59 However, concentrations can occur in individuals who take an overdose of the drug or who are slow metabolizers and develop high concentrations on what are usually therapeutic doses.221,225 Slow metabolizers are typically deficient in the cytochrome P450 enzyme, CYP 2D6, either because of genetics or because they are on a concomitant drug that substantially inhibits this enzyme (eg, fluoxetine or paroxetine).45,213
Inhibition of fast sodium channels produces stabilization of electrically excitable membranes and clinically results in the potentially life-threatening adverse effects that TCAs can have on the heart (eg, conduction disturbances) and the brain (eg, seizures).33,222,225 Thus, effects of TCAs on this SOA cause their narrow therapeutic index.
Figure 3.3 illustrates another complicating feature of the pharmacology of clomipramine and the other tertiary amine TCAs. They are demethylated in the body to secondary amine TCAs which have a pharmacological profile different from that of the parent drug. In the case of clomipramine, this metabolite is desmethylclomipramine. Its binding affinity for the same SOAs is shown in Figure 3.3 in the bar below that for the parent drug. As can be seen, this metabolite, like all secondary amine TCAs, is considerably more potent than the parent drug as an inhibitor of the norepinephrine uptake pump and less potent as an inhibitor of the serotonin uptake pump.34 The conversion of clomipramine to desmethylclomipramine is mediated by at least two CYP enzymes, CYP 1A2 and 3A3/4,43,44 and possibly 2C19.168 Activity of these 2 enzymes can vary substantially among individuals and even within the same person because these enzymes can be induced and inhibited by environmental factors, including concomitant medications taken by the individual (discussed further in Sections 7 and 8).
If inhibition of the serotonin uptake pump is critical to the desired clinical effect of clomipramine (eg, efficacy in some forms of major depression and in obsessive-compulsive disorder), then a patient may fail to respond because s/he develops higher levels of the metabolite as opposed to the parent drug. Conceivably, a patient who had responded might lose efficacy if exposed to an environmental agent capable of inducing CYP 1A2 or 3A3/4 after being stabilized on what had previously been an optimal dose of clomipramine. While the physician can increase the dose of clomipramine sufficiently to achieve high enough levels of the parent drug to produce the necessary inhibition of the serotonin uptake pump, the dose may have to be so high that effects of the metabolite on other SOAs can become clinically meaningful, causing nuisance and/or serious adverse effects. Thus, Figure 3.3 illustrates the potential problems inherent in having an active metabolite with a pharmacological profile which is meaningfully different from the parent compound.
Figure 3.3 also shows the binding affinities for all of the SSRIs. The most potent action of each SSRI is the inhibition of the serotonin uptake pump. Each has a substantially higher affinity for this SOA than for any other site shown. Stated in another way, the SSRIs as a group show a clinically meaningful separation or selectivity for the serotonin uptake pump versus any of the other SOAs shown in Figure 3.3.
However, Figure 3.3 does not show the affinity of these drugs for various CYP enzymes. In the case of these enzymes, some of the SSRIs produce meaningful effects at the same concentration that they affect the serotonin uptake pump and, thus, do not show "selectivity" in terms of distinguishing between the serotonin uptake pump and such CYP enzymes; but instead, they can produce effects on both of these sites under clinically relevant conditions (discussed further in Sections 7 and 8).
What About the Effects of SSRI Metabolites?
Given the discussion of clomipramine and desmethylclomipramine, it is important to know whether SSRIs have active metabolites with a substantially different pharmacological profile than the parent drug. Table 3.8 shows the results of two in vitro studies examining the effects of some of the SSRIs and their primary metabolites. The results for clomipramine and desmethylclomipramine, illustrated in Figure 3.3, are shown for comparison purposes. There are two entries for fluoxetine and norfluoxetine because they were examined in both in vitro studies.
As can be seen in Table 3.8, the metabolites of citalopram, fluoxetine and sertraline have the same rank order of binding affinity for these various SOAs as their respective parent SSRI. Metabolites of fluvoxamine and paroxetine were not available for testing in these studies, but they are reported to not have metabolites with clinically meaningful affinity for any of these SOAs.141,198
The metabolites of citalopram and sertraline are more than 10 times less potent than the parent drug for inhibiting the serotonin uptake pump (Table 3.8). In the case of sertraline, its metabolite is 25 times less potent than the parent drug. Since this metabolite occurs in concentrations only 1.5 times higher than the parent drug under clinically relevant conditions,219 this metabolite will be expected to contribute negligibly (ie, approximately 6%) to the overall clinical pharmacology of this drug mediated by inhibition of this SOA.
| TABLE 3.8 Effect of Uptake Inhibitors and Their Metabolites In Vitro | ||||||||
| 5-HT uptake |
NA uptake |
DA uptake |
D-2 | 5-HT2 | a1 | H-1 | ACh | |
| Clomipramine1 | 1.5 | 21.0 | 4,300 | 430 | 120 | 60 | 54 | 67 |
| Desmethylclomipramine1 | 40.0 | 0.45 | 2,100 | 1,200 | 340 | 190 | 450 | 92 |
| Citalopram1 | 1.8 | 6,100.0 | 40,000 | 33,000 | 9,200 | 1,600 | 350 | 5,600 |
| Desmethylcitalopram1 | 14.0 | 740.0 | 28,000 | 53,000 | 19,000 | 1,500 | 1,700 | 14,000 |
| Didesmethylcitalopram1 | 22.0 | 1,400.0 | 11,000 | 24,000 | 16,000 | 3,400 | 11,000 | 23,000 |
| Fluoxetine1 | 6.8 | 370.0 | 5,000 | 32,000 | 2,600 | 14,000 | 3,200 | 3,100 |
| Norfluoxetine1 | 3.8 | 580.0 | 4,300 | 13,000 | 2,500 | 15,000 | 11,000 | 3,400 |
| Fluoxetine2 | 14.0 | 143.0 | 3,050 | 12,000 | 280 | 3,800 | 5,400 | 590 |
| Norfluoxetine2 | 25.0 | 416.0 | 1,100 | 16,000 | 600 | 3,900 | 11,000 | 810 |
| Sertraline2 | 3.4 | 220.0 | 260 | 11,000 | 9,900 | 380 | 24,000 | 630 |
| Desmethylsertraline2 | 76.0 | 420.0 | 440 | 11,000 | 4,800 | 1,200 | 9,000 | 1,430 |
| Note the effects of fluoxetine and norfluoxetine were measured in two different sets of studies. Data from reference 129 are in terms of inhibition concentration, 50% maximum effect (IC50), whereas data from references 34 and 66 are in terms of kinetic inhibition constant (Ki) for the uptake pumps and kinetic dissociation constant (Kd) for the receptors. | ||||||||
| References: 1129, 234, 66 | ||||||||
The reverse is true for fluoxetine. In some in vitro studies, its primary metabolite, norfluoxetine, has been found to be somewhat more potent than the parent drug at inhibiting the serotonin uptake pump (Table 3.8). Moreover, norfluoxetine levels can be twice the levels of the parent drug and persist for a substantially longer interval after discontinuation due to its slower clearance (ie, longer half-life).142,219,232 Given its affinity for the serotonin pump and its higher, longer-lived levels, norfluoxetine is an important metabolite with regard to clinical effects mediated by the inhibition of the serotonin uptake pump.
There is an important caveat to this discussion. When the statement is made that a drug does not have an "active metabolite," several questions should be asked:
With the exception of fluoxetine, none of the SSRIs have metabolites with clinically relevant effects on any of the neural sites shown in Table 3.8. However, every SSRI that has been studied has metabolites with approximately the same activity as the parent drug for the inhibition of specific CYP enzymes (for details, see Table 8.7). Hence, these metabolites are "active" with regard to inhibiting these enzymes and contributing to the effects mediated by this action (eg, the slowing of the clearance of drugs metabolized by these specific enzymes). The magnitude of the contribution by the metabolite relative to the parent drug is a function of their relative potency for the specific mechanism of action (MOA) of interest and their relative concentrations at the relevant SOA under clinically relevant dosing conditions. For example, norfluoxetine is almost 10 times more potent than the parent drug at inhibiting the CYP enzyme, 3A3/4 (Table 8.7).
| TABLE 3.9 Effect of Metabolism on the Central MOA and Half-lives of Some SSRIs | ||||
| Drug | 5-HT uptake*1 |
NE uptake1 | Half-lives*2 | Consequence |
| Clomipramine | 1.5 | 2.1 | 19 to 37 hrs2 | Loss of selectivity |
| Desmethylclomipramine | 40.0 | 0.45 | 54 to 77 hrs2 | |
| Fluoxetine | 6.8 | 370.0 | 2 to 4 days2,3 | Increased duration of action |
| Norfluoxetine | 3.8 | 580.0 | 7 to 15 days2,3 | |
| Citalopram | 1.8 | 6100.0 | 1.5 days4 | No change in selectivity or duration of action; no clinically active metabolites in terms of serotonin uptake inhibition |
| Fluvoxamine | 3.8 | 620.0 | 0.5 to 1 day5 | |
| Paroxetine | 0.29 | 81.0 | 1 day (at 20 mg/d)2 | |
| Sertraline | 0.19 | 160.0 | 1 day2 | |
| * Half-live
is a major determinant of the duration of action of a
drug. Metabolites of citalopram and sertraline are substantially weaker inhibitors of serotonin uptake than the parent drug. These metabolites also occur in concentrations either about the same as the parent drug or less. Hence, they do not contribute in a meaningful way to the effect of the drug via this mechanism of action. However, the metabolites of several SSRIs are as potent or more potent as the parent drug at inhibiting specific CYP enzymes and thus contribute to this effect (Table 8.7). |
||||
| References: 1129, 2213, 3108, 4173, 573 | ||||
Clinical Relevance of Active Metabolites
The clinical implications of the differential effects of the metabolites of clomipramine and the SSRIs on neural SOAs are summarized in Table 3.9. In the case of clomipramine, its metabolite can cause a loss of selectivity in terms of effects mediated by the inhibition of the serotonin versus the norepinephrine uptake pump. In the case of fluoxetine, its primary metabolite has the same pharmacological profile as the parent drug. In fact, norfluoxetine, relative to fluoxetine:
For these reasons, this metabolite is clinically important in terms of increasing the magnitude and the duration of clinical effects mediated by the inhibition of the serotonin uptake pump and for effects (eg, pharmacokinetic drug interactions) mediated by the inhibition of one or more CYP enzymes.
With regard to the other SSRIs, they do not have metabolites with sufficient activity at any known neural SOAs to alter or contribute to the magnitude or duration of any psychiatric effects produced by the parent drug. However, the primary metabolites of citalopram, paroxetine and sertraline, like fluoxetine, have similar potency to their respective parent drug in terms of the effects on specific CYP enzymes (for details, see Section 8). The primary metabolites of fluvoxamine have not been adequately studied to comment about this SSRI in this respect.
Conclusion
Understanding the rational development strategy that have
been used to produce the SSRIs lays the foundation for understanding
their basic neuropharmacology and why it differs from TCAs.
This knowledge also explains why the SSRIs are alike in so
many ways and also why the differences in their pharmacokinetics
and effects on CYP enzymes have become distinguishing characteristics
among these drugs.