Clinical Pharmacology of SSRI's 5 - How SSRIs as a Group Are Similar
The rational drug discovery used to produce the selective
serotonin reuptake inhibitors (SSRIs) explains why they are
similar as a class in so many ways (Table
5.1) as well as why they differ as a class from tricyclic
antidepressants (TCAs). As in the preceding chapter, a summary
of the similarities among the SSRIs using the STEPS approach
will be given rather than an exhaustive review.
TABLE 5.1 Common
Features of SSRIs With Regard to the Treatment of Major
Depression
Flat-dose antidepressant-response curve
Equivalent antidepressant efficacy at their usually
effective, therapeutic dose: 40 mg/d for citalopram,
20 mg/d for fluoxetine and paroxetine, and 50 mg/d
for sertraline*
Similar efficacy when used on maintenance basis
to prevent relapses
Usually effective, minimum dose for each SSRI produces
approximately 60% to 80% inhibition of serotonin uptake
Benign adverse effect profile compared to TCAs
* Comparable data
from fixed-dose studies not available for fluvoxamine.
Ideally, this chapter would be based
on data from studies in which patients were randomly assigned
to one of several treatment arms: one for each SSRI at their
respective, comparable antidepressant doses and one for a
placebo. Ideally, these studies would contain an adequate
number of patients randomly assigned to each of these discrete
treatment arms to determine whether there are meaningful differences
with regard to safety, tolerability and efficacy. Since there
are 5 SSRIs, such studies would have at a minimum 6 treatment
groups (ie, each SSRI and the placebo arm). Most researchers
would probably not want to gamble on doing such a study with
only 1 dose per drug because of the criticism that the dose
chosen for a given SSRI was not the truly comparable dose
for that drug and thus biased the study outcome relative to
that drug. Instead, they would have more than 1 dose arm for
each SSRI. However, each dose added per drug would multiply
the number of groups needed for the study and thus its size
and complexity. To adequately power these studies to test
for expected differences between these different drugs would
require hundreds of patients per group. Given these considerations,
it should not be surprising that such studies do not exist.
Moreover, such studies will almost undoubtably never be done
for quite practical reasons of cost and logistics.
Since the ideal data does not exist for the purposes of
this book, the next best approach is taken by relying on summary
data from comparable studies of rigorous design. To be included,
the data has to come from double-blind, placebo-controlled,
adequately powered studies. The presence of a placebo group
permits comparisons to be drawn across the studies by comparing
the drugs on the basis of placebo-adjusted differences in
such outcome variables as adverse effects and efficacy. In
some instances, comparable data has not been published for
all of the SSRIs and, therefore, some SSRIs cannot be included
in the analysis. Those instances are pointed out in the text.
Safety
There are multiple facets to this broad heading, including:
Therapeutic index
Long-term safety
Risk of pharmacodynamically and pharmacokinetically mediated
drug-drug interactions
The available data indicate that the SSRIs as a group are
remarkably similar in all of these ways with the exception
of pharmacokinetically mediated drug-drug interactions. Given
the complexity of that topic and the fact that it is a major
distinguishing characteristic of these otherwise quite similar
drugs, it is discussed at length in Sections 7
and 8.
Since all SSRIs have been designed to avoid affecting fast
sodium channels in contrast to TCAs, they all have a wide
therapeutic index (ie, the gap between the effective dose
and a potentially toxic dose). They do not affect intracardiac
conduction.36,100,165
Patients have survived overdoses of each of the SSRIs that
were many times their usually effective antidepressant doses
without serious toxicity including:
No arrhythmias
No disturbance of blood pressure
No seizures
No coma
No respiratory depression
All of these adverse effects do occur with overdose of TCAs
as little as 5 times their therapeutic doses.225
In most instances of an overdose of only an SSRI, there is
no need for medical intervention beyond observation and addressing
the reasons for the overdose.36,100,165
Drug-drug interactions, whether pharmacodynamically or pharmacokinetically
mediated, are a safety issue with any drug
since polypharmacy is a common clinical practice particularly
in the most fragile patients (ie, the elderly and those with
multiple medical illnesses). As mentioned above, pharmacokinetically
mediated drug-drug interactions with SSRIs will be the subject
of a latter section; however, this phenomenon will be briefly
mentioned here since there is overdose risk with these drugs
when they are taken in combination with other drugs.
Overdoses in the form of a suicide attempt are typically
done with more than one drug.36,100,165
SRI-induced inhibition of a specific cytochrome P450 (CYP)
enzyme can affect the toxicity resulting from such an overdose
in two ways due to inhibition of the CYP enzyme which mediates
the metabolism of the concomitant drug. If the concomitantly
ingested drug normally has extensive first pass metabolism
dependent on that enzyme, then SSRI-induced inhibition of
that enzyme should increase the bioavailability of the other
drug and thus increase the toxicity of the overdose. The inhibition
of the enzyme should also delay the clearance of the other
drugs and thus increase the duration of their toxicity. An
increased duration will involve longer care and potentially
an increased risk of time-dependent sequelae such as intercurrent
infection in an overdose patient with compromised ventilation
due to respiratory depression. One example would is an overdose
involving a TCA and an SSRI that is capable of causing substantial
inhibition of the metabolism of TCAs (eg, fluoxetine- or paroxetine-induced
inhibition of CYP 2D6) (refer to Section
8 for details).
There can be many other examples involving overdoses with
a wide range of drugs from benzodiazepines to cardiovascular
drugs to narcotics. This scenario is based on the known pharmacology
of these drugs and pharmacokinetic principles, but to date
there are no studies which have tested whether this scenario
meaningfully affects clinical outcome in
such multiple drug overdoses.
A reduced risk of pharmacodynamically mediated drug-drug
interactions is a class advantage of SSRIs over TCAs as discussed
in Section 4. Nonetheless, SSRIs
can have pharmacodynamically mediated adverse drug-drug interactions,
primarily with drugs that also affect serotonin mechanisms
of actions (MOAs). Such interactions are a class issue common
to all of the SSRIs and a direct result of the MOA they were
designed to share (ie, the inhibition of serotonin uptake).
The most serious of these adverse interactions is the central
serotonin syndrome that can occur when monoamine oxidase inhibitors
(MAOIs) are combined with SSRIs.266
Minor variants of this syndrome in terms of the number of
symptoms, their severity, and their duration can occur when
a variety of serotonin active drugs (eg, lithium, busiprone)
are added to SSRIs or MAOIs.
This pharmacodynamically mediated drug interaction results
from the combined indirect serotonin agonism caused by both
the inhibition of serotonin degradation produced by the MAOI
and the inhibition of serotonin uptake produced by the SSRI.
Together, these two actions can create a potentially catastrophic
dysregulation of a variety of basic brainstem mechanisms,
regulated by the central serotonin neural system and produce
a syndrome consisting of:
The central serotonin syndrome produced by the combined use
of SSRIs and MAOIs is particularly dangerous because of its
severity and duration due to the persistent action of MAOIs.
The duration may also be increased the longer
the half-life of the SSRI. In severe cases, this interaction
can be fatal.
To avoid this interaction, SSRIs should not be started until
2 weeks after the discontinuation of MAOIs to allow for replenishment
of MAO activity. Similarly, MAOIs should not be started until
there has been virtual full washout of the SSRIs, which takes
2 weeks for all of the SSRIs except fluoxetine. A minimum
of 5 weeks is recommended for fluoxetine if the daily dose
was 20 mg/day, and longer if the daily dose was higher, due
to the nonlinear pharmacokinetics of fluoxetine (see Section
6).
Tolerability
While the SSRIs as a class differ from the TCAs in terms
of avoiding a number of adverse effects that are mediated
by the blockade of histamine, acetylcholine, and a1-adrenergic
receptors, as well as fast sodium channels, all SSRIs as a
class produce adverse effects that are the result of the MOA
they are designed to share (ie, indirect serotonin agonism
by inhibiting the serotonin uptake pump).
The adverse effects produced by a drug can be determined
in several ways.211
The most common is by determining what adverse complaints
or physiological effects are seen to a statistically significant
greater degree in the drug-treated group versus a parallel
placebo-treated group. Table 5.2 presents
the data on this issue for 4 of the SSRIs: fluvoxamine, fluoxetine,
paroxetine and sertraline. The data in this table is from
the double-blind, placebo-controlled clinical trial databases
with these 4 SSRIs. The presence of the placebo control allowed
for a comparison of the placebo-adjusted rates of specific
adverse effects for these 4 SSRIs (ie, the incidence rate
of each specific SSRI minus the incidence rate that occurred
on its parallel, double-blind, placebo control). The reader
who is interested in a further discussion
of this approach is referred to a recent paper on this subject.211
Unfortunately, comparable data for citalopram is not available
for this analysis. Hence, comparative statements about the
rate of specific adverse effects cannot be made about citalopram.
Nevertheless, citalopram has been reported to produce the
same type of adverse effects now known to be caused by serotonin
uptake inhibition.173
As can be readily seen in Table 5.2,
there are only modest differences in the incidence rates for
various adverse effects for these different SSRIs consistent
with how these drugs are developed. Since data in this table
are from the clinical trials databases with these drugs, it
represents the average incidence on the various doses of that
SSRI used in its clinical trials. The incidence and severity
of many of these adverse effects are dose-dependent as seen
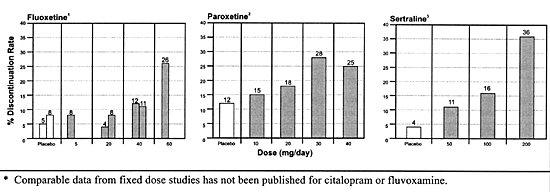
in Figure 5.1, which shows the discontinuation
rate due to adverse effects as a function of dose for the
3 SSRIs which have
published fixed-dose studies.
TABLE 5.2
Comparison of the Placebo-adjusted Incidence Rate
(%) of Frequent Adverse Effects for SSRIs*
Item
Fluoxetine
(n=1730, n=799)1
Fluvoxamine
(n=222, n=192)1
Paroxetine
(n=421, n=421)1
Sertraline
(n=861, n=853)1
Headache
4.8
2.9
0.3
1.3
Nervousness2
10.3
7.6
4.9
4.4
Tremors
5.5
6.1
6.4
8.0
Insomnia
6.7
4.0
7.1
7.6
Drowsiness3
5.9
17.2
14.3
7.5
Fatigue4
5.6
6.2
10.3
2.5
Dizziness/lightheadedness
4.0
1.3
7.8
5.0
Vision disturbances
1.0
0
2.2
2.1
Nausea
11.0
25.6
16.4
14.3
Diarrhea
5.3
0.4
4.0
8.4
Dry mouth
3.5
1.8
6.0
7.0
Anorexia
7.2
8.6
4.5
1.2
Dyspepsia
2.1
3.2
0.9
3.2
Frequent micturation
1.6
0.6
2.4
0.8
Constipation
1.2
11.2
5.2
2.1
Sweating
4.6
1.3
8.8
5.5
Respiratory5
5.8
1.3
0.8
0.8
Palpitations6
0.1
NA
1.5
1.9
Urinary retention7
NA
2.7
0.9
* Data for
fluoxetine, paroxetine and sertraline is from reference
211; data for fluvoxamine
is from reference 89.
Incidence of each respective adverse effect for
patients taking each drug minus the incidence for
each drugs parallel placebo condition.
1The
first value is the number of patients on that medication,
while the second represents those treated in the
parallel, placebo group. 2Nervousness is a composite of the following
terms: nervousness, anxiety, agitation. 3Includes somnolence, sedation. 4Includes asthenia, myasthenia, hypokinesia.
5Includes respiratory disorder, upper
respiratory infection, flu, dyspnea, pharyngitis,
sinus congestion, oropharynx disorder. 6Includes tachycardia. 7Includes micturition disorder, difficulty
with micturition, and urinary hesitancy.
NA = Not available
FIGURE 5.1 Discontinuation
Rate Due to Adverse Events as a Function of Dose
for Three SSRIs*
TABLE
5.3 Adverse Events for Each SSRI that Occurred
³ 1% More Often Than With Other SSRIs*
Fluoxetine
Fluvoxamine
Paroxetine
Sertraline
Nervousness/agitation/anxiety
Respiratory complaints
Headache
Nausea
Drowsiness
Constipation
Anorexia
Anorexia
Frequent micturition
Asthenia/fatigue
Dizziness
Sweating
Loose stools
Tremors
Dry mouth
* Placebo-adjusted
rates using the results of placebo-controlled, double-blind
studies for each SSRI respectively. Based on analysis
of data in Table 5.2. Dose-dependent
adverse effects of SSRIs which can mimic symptoms
of major depression.
NOTE: Insomnia
is another dose-dependent, adverse effect of SSRIs
and also can be a symptom of major depression. Insomnia
is not shown above because its incidence as an adverse
effect is virtually identical for fluoxetine, paroxetine
and sertraline, but is lower for fluvoxamine (Table
5.2).
Given the dose-dependent nature of many of SSRI-mediated
adverse effects, a specific SSRI can be at a disadvantage
in Figures 5.1 and 5.2
and Table 5.3 if the doses used predominantly
in its clinical trials development were higher than its usually
effective, therapeutic dose while the other SSRIs were dosed
closer to their usually effective dose. In fact, the use of
higher than necessary doses was the case for virtually all
of the SSRIs, particularly in the early phases of their clinical
trials. The reason is that the relatively benign adverse effect
profile of the SSRIs in comparison to the TCAs allowed the
investigators to titrate to the highest doses permitted in
the ascending dose design studies that were typically used
in the early studies with these drugs. This dose issue is
one of the limitations of this approach in contrast to having
data from the ideal study described at the beginning of this
chapter. Nonetheless, the data in Table
5.2 is state-of-the-art and fortunately the dosing issue
is somewhat mitigated by the fact that overdosage during the
clinical trials was virtually universal with these drugs.
With this caveat in mind, Table 5.3
lists specific adverse effects that have at least a 1% higher
incidence rate on a specific SSRI in comparison to the other
3.
The SSRIs as a class also produce a variety of sexual dysfunction
adverse effects, including anorgasmia and decreased libido
(Table 5.4). Although an analysis of
the clinical trial database for each SSRI suggests that fluvoxamine
and fluoxetine are less likely to produce these effects than
paroxetine and sertraline, clinical experience suggests that
all SSRIs produce a comparable rate at their usually effective,
minimum antidepressant dose.130,280,296
One possible reason for the lower rates in the clinical trial
databases for fluvoxamine and fluoxetine is that these two
SSRIs were the first to be extensively studied and that unfamiliarity
with this adverse effect may have contributed to an under-reporting
of its occurrence. Again, the ideal study described at the
beginning of this chapter would more convincingly answer this
question; in the interim, clinicians will have to assess this
matter using the available data and their clinical experience.
Clearly, sexual dysfunction is an unintended effect that can
apparently be produced by serotonin uptake inhibition.
Of importance, a number of the dose-dependent adverse effects
produced by the SSRIs can mimic clinical depression (Tables
5.3 and 5.4).
These include:
Nervousness/agitation/anxiety
Drowsiness or daytime tiredness
Anorexia
Fatigue
Sexual dysfunction, such as decreased libido
TABLE 5.4 Placebo-adjusted
Incidence (%) of Various Forms of Sexual Dysfunction on
Four SSRIs1
Adverse Effect
Fluoxetine
(n=1730, n=799)2
Fluvoxamine
(n=222, n=192)2
Paroxetine
(n=421, n=421)2
Sertraline
(n=1033, n=1033)2
Abnormal ejaculation/orgasm3
1.4
12.9
13.3
Other male gender disorders4
10.0
Decreased libido
1.6
3.3
Sexual dysfunction (male)
1.9
Sexual dysfunction (female)
1.5
Female genital disorder5
1.8
Menstrual disorder6
0.5
Painful menstruation
0.5
Incidence is based on gender whenever appropriate.
Placebo incidence on average for each of the above
categories is under 0.5%.
The first value is the number of patients on that
medication, while the second represents those treated
in the parallel, placebo group.
Incidence based on number of male patients.
Includes anorgasmia, delayed orgasm, erectile dysfunction,
impotence, and sexual dysfunction.
This fact has several implications. First, the emergence
of such adverse effects at higher doses may in part account
for the fact that the antidepressant response rate to the
SSRIs tends to be lower on average at doses higher than the
usually effective, minimum dose based on the result of fixed-dose
studies (Figure 5.2). Second, physicians
may misinterpret the emergence of such adverse effects as
the need for higher doses and thus increase the dose unwittingly,
worsening the situation. The late emergence of these effects
as a result of gradual drug accumulation may be interpreted
as the drug losing its effectiveness. This phenomenon is one
reason the physician should ensure that the patient has had
an adequate trial on the usually effective, minimum dose of
each SSRI. For the same reason, the physician may also wish
to consider a dose reduction rather than increase if the patient
initially appeared to respond and then has a recurrence of
symptoms. This phenomenon also is relevant to the potential
role of therapeutic drug monitoring (TDM) with SSRIs which
is discussed later in this section.
Efficacy - Acute
The major indication for the use of SSRIs is the treatment
of major depression. There are 3 types of efficacy that are
important in this condition:
Acute amelioration of the depressive syndrome
Maintenance of that improvement during the vulnerable
period for a relapse
Prophylactic treatment to prevent the occurrence of a
new episode92,150,151,233
There is a variable amount of data for the various SSRIs
on the first two uses and no systematic data for any of them
on their ability to prevent recurrent episodes for a period
greater than 1 year. The comparable data existing for these
2 uses in major depression does not reveal any difference
among the SSRIs in terms of either the induction
of acute response or maintenance of that response for a period
up to 1 year (Table 5.5).
Before reviewing that data, it is important to note that
the efficacy of SSRIs is not limited to major depression but
extends to several other conditions including obsessive-compulsive
disorder, panic disorder, and even conditions such as premature
ejaculation.156,199
The efficacy of these drugs in these other conditions appears
to be due to their indirect serotonin agonism via serotonin
uptake inhibition and hence is probably a class phenomenon.
Different SSRIs have approval for several of these other indications
in different countries, others have applications pending for
such approval, and still others are in clinical testing in
hopes of obtaining data that will support application for
formal labeling for these conditions. Fluvoxamine is an example
of vagaries of such approvals. It is formally labeled in the
US for the treatment of obsessive-compulsive disorder but
not for major depression even though it has that indication
in many other countries. Since major depression is the principal,
clinical use of these drugs and has the largest amount of
comparative data, it will be the focus of this discussion
of their comparative efficacy.
Two approaches were taken to compare the efficacy of the
different SSRIs in terms of the acute relief of depressive
symptoms. The first was a meta-analysis of the double-blind,
placebo-controlled studies and the second was a comparison
of the results of placebo controlled, fixed-dose studies.
The latter is critical to determining whether there is a difference
among the drugs in terms of their efficacy at the usually
effective, minimum dose. Of necessity, these comparisons had
to be limited to the
SSRIs for which published data existed from such studies.
TABLE 5.5 SSRI
versus Placebo: Response Rate and Relapse Rate
Response
Rate*(%)1
P
Value
Duration1
(weeks)
Relapse
Rate (%)2-4
P
Value
SSRI
Placebo
Difference
Placebo
Drug
Difference
Fluoxetine
60
33
27
< 1013
52
57
26
31
< 0.01
Fluvoxamine
67
42
25
< 102
NA
NA
NA
NA
NA
Paroxetine
65
36
29
< 1014
52
43
16
27
< 0.01
Sertraline
79
48
31
< 1011
44
46
13
33
< 0.001
The meta-analysis
for response rate did not include citalopram. A relapse
prevention study has been done with citalopram, but lasted
24 weeks rather than one year. Its results are discussed
in the text. A relapse prevention study has not been published
for fluvoxamine.
* Response defined
as at least 50% decrease in depression symptom severity,
as measured using a standard instrument. Duration of
maintenance treatment follow-up.
It was possible to do the meta-analysis with 4 of the 5 SSRIs:
fluvoxamine, fluoxetine, paroxetine and sertraline.226
The results of such studies with citalopram were
not published in sufficient detail at the time of this meta-analysis
to permit that SSRI to be included. Based on the results of
this meta-analysis, each SSRI produces approximately a 60%
overall response rate (ie, at least a 50% reduction in symptoms
as a result of treatment) and a 30% higher response rate than
a parallel, placebo control. This meta-analysis suggests that
approximately the same percentage of patients with major depression
respond to approximately the same degree to each of these
4 SSRIs. The data that has been published on citalopram suggests
its efficacy is comparable.174,177
Double-blind, placebo-controlled, fixed-dose studies are
the only way to determine the optimal dose of a drug and what
is a comparably effective dose for different drugs in the
same class (ie, having the same MOA mediating that outcome).
Such studies have been done with all of the SSRIs, but the
results have not been presented yet for the citalopram or
fluvoxamine studies. Therefore, this analysis is of necessity
limited to the studies with fluoxetine, paroxetine and sertraline
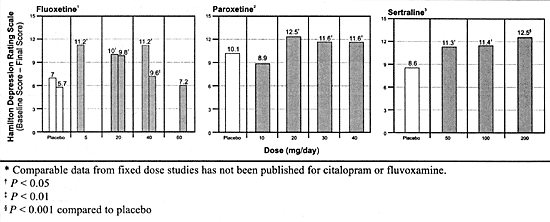
(Figure 5.2).
Based on available studies, each of these 3 SSRIs have a
flat-dose antidepressant-response curve meaning that they
produce approximately the same average response rate at each
dose above their usually effective, minimum dose over their
clinically relevant dosing range (Figure
5.2). Based on these studies, the usually effective, minimum
dose for fluoxetine and paroxetine is 20 mg/day and for sertraline
50 mg/day. Paroxetine was the only 1 of the 3 to include an
ineffective dose, 10 mg/day.80
In 1 study, fluoxetine (5 mg/day) was as effective as 20 mg/day
on the Hamilton Depression Rating Scale but was not effective
on other measures. This lack of robust effectiveness and the
limited studies with fluoxetine 5 mg/day has been the basis
for concluding that 20 mg/day is the optimal dose.6
The conclusion that 50 mg/day of sertraline is its
optimal dose based on its fixed-dose study is further supported
by a review of its other efficacy studies.229
If anything, the response rate in the fixed-dose clinical
trials tends to be lower at higher doses of fluoxetine and
paroxetine. As mentioned above, one reason for this result
may be the emergence of dose-dependent, adverse effects which
mimic symptoms of major depression. Consistent with this explanation,
the discontinuation rate due to adverse effects increases
at higher doses for all 3 of these SSRIs (Figure
5.1).
Given the delayed onset of antidepressant response seen
with SSRIs and other antidepressants, an increase in dropouts
at higher doses will also bias against the response rate at
higher doses since patients will be prone to discontinuing
the trial before they may reasonably be expected to respond.
Regardless of the reason, the decrease in the average magnitude
of the antidepressant effect for fluoxetine and paroxetine
reinforces the recommendation to ensure that the patient has
had an optimal trial on the usually effective dose of the
SSRIs before attempting a dose increase unless the physician
uses a TDM approach to judge the adequacy of the dose in the
patient as discussed later in this chapter.
The usually effective dose of fluvoxamine and citalopram
have not been convincingly established due to the absence
of published data from appropriately designed and executed
fixed-dose studies. Nonetheless, a meta-analysis of the placebo-controlled
studies with citalopram has been published in 2 separate publications.174,177
The results of this meta-analysis suggest that citalopram
also has a flat-dose antidepressant-response curve in terms
of antidepressant efficacy.
Nevertheless, this meta-analysis is not an adequate substitute
for publication of the fixed-dose studies. Its interpretation
is compromised by the fact that the studies involve different
criteria for subject selection, different designs,
and different scales. Additionally, it is based on the data
from only approximately 60% of the patients who were assigned
to the citalopram treatment arms (ie, a selected group of
patients who stayed on treatment for at least 4 weeks). A
case was made using this meta-analysis that 20 mg/day of citalopram
is the usually effective dose in patients who do not have
severe and/or melancholic major depression. However, the published
results of the 1 fixed-dose study with citalopram showed that
40 mg/day was statistically superior to placebo at weeks 3,
4 and 6 and superior to 20 mg/day at weeks 4 and 6 (p
< 0.05), while 20 mg/day was not statistically superior
to placebo at any time.178
In contrast to the meta-analysis, the analysis of this fixed-dose
study used the more conservative and more generally accepted
"last observation carried forward" approach. Until
the results of the other fixed-dose studies with citalopram
are adequately presented, the results from the published,
fixed-dose study serves as the basis for concluding that 40
mg/day is the usually effective, minimum dose for citalopram
as shown in Table 3.7.
Returning to the fixed-dose studies with fluoxetine, paroxetine
and sertraline, the magnitude of the antidepressant effect
(ie, the absolute reduction in depressive symptoms as quantitated
using the Hamilton Depression Rating Scale) was also virtually
identical for these 3 SSRIs at their usually effective, minimum
dose (Figure 5.2). These findings suggest
that each of these drugs treats approximately the same percentage
of patients to approximately the same extent at their respective
minimally effective doses. Consistent with this finding is
the fact that each of these SSRIs at these doses produces
approximately a 70% to 80% inhibition of serotonin uptake
using the platelet as a surrogate marker for the effect on
the central serotonin neurons (Table
3.7). The plasma concentration of each SSRI at comparable
antidepressant doses are consistent with
the concentrations predicted to be needed to produce this
magnitude of uptake inhibition based on the in vitro
IC50 data.
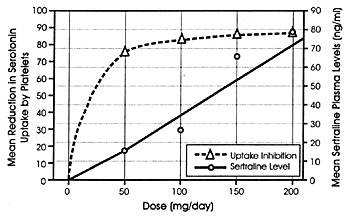
Figure 5.3 further illustrates that
the major effect on serotonin uptake inhibition with SSRIs
occurs at the lower end of their clinically relevant dosing
range using data from studies with sertraline as a representative
SSRI.223 In this
study, normal volunteers were randomly assigned to 1 of 4
fixed-doses of sertraline. The plasma levels that they achieved
and the degree of serotonin uptake inhibition that occurred
on each of the 4 doses was measured. The platelet was used
as a surrogate in lieu of measuring the effect of the drug
on central serotonin neurons. As is apparent in Figure
5.3, 80% of serotonin uptake inhibition was achieved at
plasma levels produced by the 50 mg/day dose. While both the
plasma levels of the drug and the degree of serotonin uptake
inhibition increased as expected with the higher fixed-doses,
the higher levels at 200 mg/day were associated with only
an additional 8% increase in serotonin uptake inhibition further
reflecting the fact that majority of the effect had already
occurred and the plateau portion of the dose-response curve
had been reached. The fact that this dose-response curve is
so similar to the dose-response curve for the antidepressant
efficacy of the drug (ie, that is relatively flat above 50
mg/day) provides an heuristic explanation consistent with
the presumed MOA for both effects being the result of the
effect of sertraline on the serotonin uptake pump. The relationship
illustrated in Figure 5.3 is not unique
for sertraline but is simply illustrative of curves that can
be drawn for any of the SSRIs which have been so studied.
This phenomenon is again consistent with how these drugs
were rationally developed.
FIGURE 5.3 Relationship Between
Daily Dose of Sertraline, Mean Plasma Levels of Sertraline,
and Mean Reduction in Serotonin Uptake by Platelets After
14 Days of Drug Administration at One of Four Fixed Doses
All of the above observations raise serious questions about
the widespread clinical practice of routinely using higher
doses of SSRIs before there has been an adequate trial on
their usually effective, minumum dose. The only double-blind,
adequately powered study to test whether early nonresponders
to an SSRI would benefit from a higher dose randomly assigned
patients who had not responded after 3 weeks of treatment
with fluoxetine 20 mg/day to either stay on 20 mg/day or be
treated with 60 mg/day.77
At the end of an additional 5 weeks of treatment, an equal
number of patients had responded in both groups and the time
course for response was also the same. This study is fully
consistent with the flat-dose antidepressant-response curve
and indicates that 3 weeks is not a sufficient trial to determine
that the usually effective dose of this SSRI is inadequate
for a patient. Parenthetically, fluoxetine is not the ideal
SSRI for this type of study since the long
half-lives of the parent drug and active metabolite may make
a delayed response more likely than with the other more intermediate-lived
SSRIs. Nonetheless, the data exists for this SSRI and not
the others.
The reason to adequately test the antidepressant response
to the usually effective, minumum dose is to optimize the
tolerability of the drug and to avoid unintended effects on
CYP enzymes (see Sections 7 and
8). While the effect on antidepressant
response and serotonin uptake inhibition of the SSRIs has
on average reached the plateau portion of their dose-response
curve, that is not true for their discontinuation rate due
to adverse effects (Figure 5.1) nor
for the effects of specific SSRIs on specific CYP enzymes
(eg, fluoxetine on CYP 2D6). Thus, the trade-off for using
higher doses than necessary is to increase the degree of CYP
enzyme inhibition produced, possibly the number of CYP enzymes
inhibited and number and severity of dose-dependent adverse
effects which can mimic major depression. All of these adverse
consequences can occur with higher than the usually effective
dose without improving antidepressant efficacy.
These observations also raise serious question about the
widespread clinical practice of switching from one SSRI to
another if the patient fails to respond to the first SSRI
after a reasonable therapeutic trial. Conceivably, there is
some as yet unidentified difference in the spectrum of antidepressant
activity of these drugs, but the available data is not suggestive
that a meaningful difference exists. There is certainly no
compelling double-blind, placebo-controlled data to support
this practice. It is clearly an important deficiency in our
knowledge when faced with a patient who has not responded
to a member of this class of drugs.
In addition to acute efficacy, fluoxetine, paroxetine and
sertraline have been studied in terms of their ability
to prevent a recurrence of a depressive episode in the 1-year
interval following the induction of an acute response (Table
5.5). The design of these studies is similar so that the
results can be reasonably compared: patients were brought
into remission on the SSRI and after a period of several months
of stabilization were randomly assigned in a double-blind
fashion to either remain on the SSRI or be switched to placebo.
As with acute efficacy, the results of these studies are amazingly
consistent: continued treatment with each SSRI produced an
approximately 30% reduction in the risk of relapse compared
to the parallel, placebo control in the 1-year period of follow-up
observation. A similar study was done with citalopram, but
for 6 months rather than 1 year. Like the other 3 SSRIs, citalopram
was superior to placebo having a 20.5% lower risk of relapse
at that point.179
That result is comparable to the result with the other 3 SSRIs
at the same time point in the longer studies with those drugs.
A relapse prevention study has not been published with fluvoxamine.
The expectation is that it will be similarly effective.
Therapeutic Drug Monitoring
The discussion about the flat-dose antidepressant-response
curve is not meant to imply that there are no patients who
will benefit from a dose other than the usually effective,
antidepressant dose. The phrase "on average" is
key to that discussion. Like the effect on serotonin uptake
inhibition (Figure 5.3) and specific
CYP enzyme inhibition (Figure 8.3
and 8.4), the antidepressant
effect and adverse effects of SSRIs must be concentration-dependent
although the "signal-to-noise" problems in such
research makes it difficult to show a strong correlation between
these clinical effects and plasma drug levels.
Since the plasma levels achieved on the same dose of the
same SSRI can differ among patients, there are undoubtably
some patients who need a lower dose to achieve the concentration
that usually occurs on the usually effective, antidepressant
dose and, conversely, some patients need a higher dose to
achieve the same levels. The problem is that it is obviously
difficult to detect this fact using dose alone.
One approach to this problem is simply careful dose titration
based on clinical assessment of response. However, there are
several problems with this approach. First, there is no compelling
data about what is an adequate duration to know that the usually
effective dose is wrong for that specific patient. The only
data on this issue indicates that 3 weeks is an inadequate
trial for fluoxetine, but not what is an adequate trial for
this or any other SSRI. Second, the clinician would have to
be able to distinguish between dose-dependent, adverse effects
that mimic depressive symptoms and the depressive symptoms
themselves. The placebo response rate in clinical trials does
not suggest that even experienced clinical investigators can
consistently determine when a response is specifically treatment
related. For these reasons, TDM can have a role in deciding
whether a specific patient needs a dose other than the usually
effective, minimum dose.
A few general comments may be helpful to put this discussion
in perspective. Unlike TCAs, TDM with SSRIs will almost undoubtably
never be a standard-of-care issue. The avoidance of serious
toxicity is the overriding rationale for TDM. That is not
an issue with the SSRIs in contrast to the TCAs. Due to their
narrow therapeutic index, their potentially life-threatening
toxicity, and wide interindividual variability in clearance,
some patients on conventional antidepressant doses of TCAs
can experience serious adverse consequences (eg, delirium,
seizures, arrhythmias).221
The avoidance of such toxicity primarily in patients
who are deficient in CYP 2D6 function either due to genetics
or concomitantly prescribed drugs (eg, fluoxetine, paroxetine)
is the reason for obtaining a plasma level once early in the
course of treatment with a TCA to permit a rational dose adjustment
if needed.
A second and less pressing reason to use TDM is to increase
efficacy and tolerability by adjusting the dose upward in
patients who have rapid clearance to increase efficacy and
downward in those with slow clearance to improve tolerability
and thus efficacy.221
This reason would be applicable to drugs like the SSRIs which
have a sufficiently wide therapeutic index such that serious
toxicity is not a concern, but nevertheless can produce a
greater increase in adverse effects than an increase in efficacy
on average at higher doses (Figures 5.1
and 5.2), especially since some of those
adverse effects can be confused with lack or loss of efficacy
(Table 5.3).
The problem is establishing the plasma levels of the various
SSRIs that will produce the optimal balance of antidepressant
efficacy and tolerability. One approach that has been used
is to try to correlate plasma levels of the various SSRIs
with antidepressant response in clinical trials. Such studies
have been published with all of the SSRIs and have all failed
to show a relationship between drug concentration and antidepressant
response: citalopram,31,79
fluoxetine,232,142
fluvoxamine,37,140
paroxetine,158,269
and sertraline.223
That is not surprising for many reasons including the "signal-to-noise"
problem created by placebo response and the fact that these
studies used doses at or above the usually effective, minimum
dose. In other words, they examined the plateau portion of
the dose-response curve where one would predict that there
would be no relationship between concentration and response.
The concentration-dependent nature of antidepressant response
with the SSRIs is apparent from the flat-dose antidepressant-response
relationship and the fact that it parallels
the dose- and concentration-dependent effect on serotonin
uptake inhibition (Figure 5.3). This
relationship is consistent with a minimum threshold concentration
that produces approximately 70% serotonin uptake inhibition
and exerts a step-function effect on antidepressant response.
However, a study demonstrating such a relationship will have
to focus on doses which produce concentrations both below
this level and somewhat above, in contrast to the studies
that have been done. Such a study will also have to involve
hundreds of patients to overcome the "noise" in
current antidepressant clinical trials research. The interested
reader is referred to a previous paper that discusses the
technical problems of doing such research in psychiatry.230
The ideal TDM study with the SSRIs described above will
most likely never be done for cost and logistical reasons.
There is no compelling incentive on the part of any government
or private research sponsor to demonstrate the concentration-dependent
nature of antidepressant response to SSRIs. In fact, the manufacturer
may actually have a disincentive out of concern that the demonstration
of such a relationship might be used to suggest that TDM is
necessary with their medication and thus
create a perception that may adversely impact the clinical
acceptance of the drug.
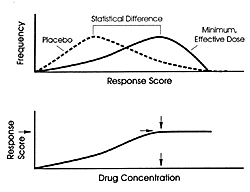
FIGURE 5.4 Estimated Minimum
Effective Drug Concentration: Average Plasma Drug Concentration
Achieved in the Group Treated With the Minimum, Effective
Dose
In the absence of data from such an ideal TDM study, the
fixed-dose studies can be used to estimate the optimal plasma
level range as illustrated in Figure 5.4.
Fixed-dose studies take advantage of group difference to establish
the superiority of the drug over placebo. As illustrated in
this figure, some patients will do as well on placebo as the
best patient does on the drug at any dose, and some patients
on the drug will do as poorly as any patient on placebo. In
other words, there is complete overlap in the range of the
response between the two groups. That is what is meant by
the term, "noise," in these studies. The ability
to show a difference between drug and placebo is based on
the fact that the curve for the drug-treated group is skewed
to the responder side, whereas the curve of the placebo-treated
group is skewed to the nonresponder side to a statistically
significant extent The usually effective, minimum dose is
the lowest dose that separates the drug from placebo. In the
case of the SSRIs, it is also the best dose for most patients
(Figure 5.2).
The usually effective dose defines an expected concentration
range (Figure 5.4) based on the variability
of the clearance of the drug in different patients. Since
this dose on average separates drug treatment from placebo,
the concentration that on average is achieved by this dose
must also separate the drug from placebo. This concentration
then is an estimate of the minimum desired concentration.
Since higher doses on average increase
adverse effects more than efficacy, the average plasma levels
produced by those doses define the upper threshold where generally
nuisance adverse effects outweigh additional therapeutic benefit.
It would be ideal to have a low dose in the fixed-dose study
that does not separate the drug from placebo because the concentration
achieved by that dose will more convincingly establish the
minimum threshold concentration. Such data exists for paroxetine
(10 mg/day)80 and
possibly for citalopram (£ 20 mg/day).178
If that has not been established, then one does not know whether
a lower threshold will actually be better on average in terms
of the balance between efficacy and tolerability.
This information can help the clinician in the decision-making
process. A patient may have failed on the usually effective,
minimum dose because s/he is not responsive to this MOA or
because plasma concentration of the drug is below the usually
effective threshold due to an unusually rapid clearance of
the drug in that patient. TDM can be used to provide information
relevant to the latter possibility. If TDM reveals substantially
lower plasma drug levels than would normally be expected for
patients on this dose, the problem may be noncompliance or
rapid clearance. In the former case, the physician could visit
with the patient and determine what is leading to compromised
compliance. In the event of rapid clearance, a trial of a
higher dose will be more rational than switching to another
SSRI or to another class of antidepressants. The goal will
be to ensure that each patient receives a trial of a dose
which will result in that patient achieving plasma drug levels
that are usually therapeutic for that drug. If the patient
has a level considerably above the range produced by the usually
effective, minimum dose due to unusually slow clearance, then
a dose reduction may be warranted to determine whether the
less than optimal response is due to adverse effects that
are mimicking depressive symptomatology.
The goal of this TDM approach is to aid rather than to dictate
the dose adjustment. Such TDM data would have to be considered
by the physician along with other factors specific to that
patient such as clinical assessment of efficacy and tolerability.
As stated above, TDM is clearly not a necessity with SSRIs.
Their wide therapeutic index means that clinicians can titrate
the dose over large ranges without concern about serious toxicity.
Some physicians will, therefore, never use TDM with these
drugs. Others may use this approach in nonresponders. A few
might even wonder whether this TDM approach might have sufficient
clinical and cost-effectiveness advantages to use it early
in treatment as a routine aspect of treatment. The goal in
this latter approach would be to reduce the time needed to
determine whether the usually effective, minimum dose in that
patient is producing the usually expected plasma levels in
the patient. The potential advantages will be reducing the
duration of the illness by being able to more rapidly make
dose adjustments and by not erroneously concluding that the
patient is not responsive to the drug when the problem is
unusually rapid or slow clearance of the drug.