|
|
Clinical Pharmacology of SSRI's
6 - What Are the Clinically Relevant Pharmacokinetic Differences Among SSRIs? |
|
|
|
|
|
While the serotonin selective reuptake inhibitors (SSRIs)
were rationally developed to be similar in terms of avoiding
effects on several neural sites of action (SOAs), they were
not designed to be similar with regard to their pharmacokinetics.
Not surprisingly, this area is one where there are clinically
meaningful differences among these drugs.
Concentration-dependent Effects
of SSRIs
The concentration-dependent effects of SSRIs are summarized
in Table 6.1. Chapter 5 reviewed the
data on the concentration-dependent nature of the effects
of SSRIs in terms of the inhibition of the serotonin uptake
pump, their antidepressant efficacy, and the incidence and
severity of their serotonin-mediated adverse effects. The
concentration-dependent inhibition of specific CYP enzymes
by specific SSRIs will be presented in Sections 7
and 8. The concentration-dependent
nature of these effects is the basis for the clinical importance
of the pharmacokinetic differences among
SSRIs (Table 6.2).
| TABLE 6.1 Concentration-Dependent
Effects of SSRIs |
- Serotonin uptake inhibition (eg, platelets)
- Antidepressant efficacy
- Incidence and severity of serotonin-mediated adverse
effects
- Competitive inhibition of specific CYP enzymes
|
The only pharmacokinetic parameters shared by all the SSRIs
is that they are relatively slowly, but completely, absorbed
from the gut (ie, time to peak plasma concentration is 3 to
8 hours) and have large apparent volumes of distribution.213
They differ with regard to:
- Their protein binding
- Their metabolism including which CYP enzymes are principally
responsible for their biotransformation
- Their half-lives
- Whether they have linear or nonlinear pharmacokinetics
over their clinically relevant dosing range
- Whether they have active metabolites
- The effect of age and specific organ impairment on their
elimination rates
Protein Binding
Fluoxetine, paroxetine and sertraline are highly protein
bound (ie, > 95%).213
In contrast, the protein binding of citalopram (50%) and fluvoxamine
(77%) is considerably less.94,199
High protein binding raises the possibility of displacement
interaction with other highly protein bound drugs. However,
SSRIs are weakly bound primarily to a1-acid glycoprotein.
Perhaps for this reason, even the highly protein bound SSRIs
have not been found to increase the free fraction of concomitantly
administered drugs that are also highly protein bound.
| TABLE 6.2 Pharmacokinetic
Parameters Relevant to the Use of SSRIs |
| Parameter |
Citalopram |
Fluoxetine |
Fluvoxamine |
Paroxetine |
Sertraline |
| Autoinhibition1 |
No |
Yes |
Weakly yes |
Yes |
No |
| Half-life (in days)2 |
1.5 |
2 to 43
(7 to 15)4
|
0.5 to 1 |
13 |
1 |
| Time to steady-state (in days) |
6 to 7 |
30 to 603,4
|
3 to 5 |
4 to 54
|
4 to 5 |
| Dose-plasma level proportionality5 |
Yes
|
Yes |
Weakly no6
|
No |
Yes |
| Active metabolite in terms
of comparable in vitro7 potency to parent drug
for inhibiting specific CYP enzymes |
Yes |
Yes |
Unknown |
Yes |
Yes |
- See Table 6.4.
- See Table 3.9.
- At their usually effective, therapeutic dose (ie,
20 mg/day), but longer at higher doses due to autoinhibition.
- Half-life (t1\2) of norfluoxetine which has virtually
the same pharmacological profile as the parent drug
and, hence, must be considered when calculating time
to steady-state, time to maximal effect, and time
to washout of effect.
- See Table 6.5.
- Based primarily on results summarized in Table
6.4; as seen in Table 6.5,
there is scant published data measuring steady-state
plasma levels at different doses for these two SSRIs.
- See Table 8.7.
|
Metabolism
While some SSRIs can competitively inhibit specific CYP enzymes,
the interaction between CYP enzymes and SSRIs is a two-way
street. All of the SSRIs undergo extensive oxidative metabolism
as a necessary step in their eventual elimination; however,
different CYP enzymes mediate the metabolism of different
SSRIs (Table 6.3). This knowledge is
important because it forms the basis for understanding many
of the other pharmacokinetic differences among the SSRIs.
It is also important in terms of knowing whether concomitantly
administered drugs can affect the clearance of specific SSRIs
and hence their efficacy and tolerability. After all, SSRIs
can be the target of pharmacokinetic drug-drug interactions
as well as being the cause of such interactions.
Information about their metabolism varies substantially
from one SSRI to another. The metabolism of citalopram and
paroxetine have been the best characterized and sertraline
is intermediate in terms of our knowledge; whereas there is
considerably less information on the metabolism
of fluoxetine and fluvoxamine, although for different reasons.
| TABLE 6.3 CYP Enzyme
Responsible for Biotransformation of SSRIs |
| SSRI |
CYP Enzyme Metabolized |
| Citalopram |
2C19 mediates initial
step, then 2D6 |
| Fluoxetine* |
2D6, partially responsible;
remainder not established |
| Fluvoxamine |
Not known |
| Paroxetine* |
2D6, principal
P450 |
| Sertraline |
3A3/4 responsible for demethylation |
|
* The inhibition of this enzyme is
responsible for nonlinear pharmacokinetics of paroxetine
and at least partially for the nonlinear pharmacokinetics
of fluoxetine.
|
As reviewed in Section 2, citalopram
is marketed as a racemic mixture and there are differences
in the enantiomers in terms of their rate of metabolism. Racemic
citalopram is metabolized by both CYP 2C19 and CYP 2D6 based
on a study comparing the steady-state levels of citalopram,
desmethylcitalopram, and didesmethylcitalopram in individuals
who were extensive metabolizers of sparteine (ie, CYP 2D6)
and mephenytoin (ie, CYP 2C19), and individuals who were poor
metabolizers of either sparteine or mephenytoin respectively.254
Both the total clearance of citalopram and the clearance of
desmethylcitalopram were significantly slower in poor versus
extensive metabolizers of mephenytoin (ie, deficient in CYP
2C19) while the demethylation clearance of desmethylcitalopram
was significantly lower in poor versus extensive metabolizers
of sparteine (ie, deficient in CYP 2D6). Both of these CYP
enzymes exhibit considerable genetic polymorphism in specific
populations: approximately 5% to 10% of white populations
in western Europe and North America are genetically deficient
in CYP 2D684,265
whereas 20% of Orientals are genetically deficient in CYP
2C19.104,152
The fact that citalopram is dependent on these 2 enzymes will
be expected to increase the interindividual variability in
plasma levels of this drug.
Although fluvoxamine was one of the first SSRIs developed,
it was developed before the technology existed to readily
identify which CYP enzyme(s) was responsible for its biotransformation.
Hence, there have been no formal studies examining which CYP
enzymes are responsible for its biotransformation. This is
unfortunate since the biotransformation of fluvoxamine is
extensive and occurs mostly by oxidation.198
Only negligible amounts of fluvoxamine
are excreted unchanged in the urine. Eleven different metabolites
have been identified in urine. However, a plot of the areas
under curve (AUCs) of fluvoxamine in nearly 100 individuals
did not reveal bimodality in distribution as would be expected
if CYP 2D6 or CYP 2C19 were the rate-limiting enzyme.73
However, smokers have a 23% reduction in fluvoxamine compared
to nonsmokers, suggesting a possible role for CYP 1A2 in fluvoxamine
metabolism.275 The
fact that fluvoxamine plasma levels are somewhat higher after
multiple doses than after a single dose is also partially
consistent with the fact that fluvoxamine can be metabolized
by CYP 1A2 since fluvoxamine inhibits this enzyme. However,
the nonlinearity in its plasma levels are not as great as
would be predicted based on its effects on other CYP 1A2 substrates
such as theophylline.
Knowledge about fluoxetine metabolism is also limited in
part because such investigations are complicated by several
factors:
- First, fluoxetine is marketed as a racemic mixture like
citalopram.
- Second, it is N-demethylated to norfluoxetine,
which is also chiral as discussed in Section
2.
- Third, fluoxetine and particularly norfluoxetine are
slowly cleared (ie, extended half-lives).
- Fourth, both inhibit multiple CYP enzymes to varying
degrees at clinically relevant concentrations and are known
to inhibit their own metabolism presumably by inhibiting
the responsible CYP enzymes. This matter is further complicated
by the fact that the relative potency for such inhibition
can vary between the enantiomers of both fluoxetine and
norfluoxetine.
Thus, the knowledge about which CYP enzyme mediates the metabolism
of fluoxetine is limited but will be summarized below to the
extent possible.
The N-demethylation of both enantiomers of fluoxetine
is probably at least in part metabolized by the same CYP enzyme(s)
based on the results of an in vitro study showing a
reasonable correlation between the rates at which human liver
microsomes catalyze this reaction.267
The R-fluoxetine was metabolized about 50% faster,
which is consistent with reports of higher levels of S-fluoxetine
in individuals on the drug.18,267
There are probably multiple CYP enzymes involved in the
metabolism of fluoxetine at different concentrations, accounting
for the nonlinear pharmacokinetics of the drug. As higher
affinity enzymes become inhibited, lower affinity enzymes
become relevant as the concentration of the drug increases.
There is evidence suggesting a role for both CYP 2D6 and CYP
3A3/4. First, the metabolism of fluoxetine cosegregates with
the CYP 2D6 polymorphism.7,24 N-demethylation
of fluoxetine positively correlates with CYP 2D6 levels in
human microsomes, but is inhibited only 20% by quinidine and
27% by antisera to CYP 2D6 and does occur in human microsomes
lacking the CYP 2D6 enzyme.18,267
The fact that the N-demethylation of fluoxetine is
autoinhibited at higher concentration suggests that this step
is mediated at least in part by another CYP enzyme which is
more weakly inhibited than CYP 2D6, such as CYP 3A3/4, 2C19,
2C9/10 or another CYP enzyme.
Paroxetine is metabolized principally to an intermediate,
which is then conjugated and eliminated.141
Two CYP enzymes mediate this reaction. There is ample evidence
that at low paroxetine concentrations, this enzyme is CYP
2D6. First, enzyme activity at low concentrations of paroxetine
cosegregates with the sparteine polymorphism and in vitro
is responsible for approximately 75% of the activity in CYP
2D6 extensive metabolizers.252,253
Second, this activity is inhibited by quinidine
and by paroxetine itself, which are both known inhibitors
of CYP 2D6.32,252
The second CYP enzyme has a much lower affinity for paroxetine.
It is responsible for 25% of the in vitro metabolism
of paroxetine in CYP 2D6 extensive metabolizers at low concentrations
but is the primary enzyme in CYP 2D6 poor metabolizers and
in extensive metabolizers at higher concentrations.253
This low affinity enzyme has not been identified but may be
inhibited by cimetidine,16,111
may decline in efficiency with age,253
and may be induced by some anticonvulsants.8
While paroxetine is principally metabolized by CYP 2D6 at
low concentrations, it also inhibits this enzyme in a concentration-dependent
manner (see Section 8). Hence,
this pathway becomes saturated at higher concentrations and
paroxetine elimination becomes dependent on the lower affinity,
but higher capacity, enzyme. The relative roles of these two
enzymes in the metabolism of paroxetine is the apparent explanation
for why paroxetine has nonlinear pharmacokinetics including
a half-life of 10 hours after a single 20 mg dose, but a half-life
of almost 24 hours after multiple doses of 20 mg/day.141
Sertraline, like the other SSRIs, is mainly eliminated by
oxidative metabolism and the dominant metabolite is N-desmethylsertraline.288
Several observations rule out a major role for CYP 2D6 in
the metabolism of sertraline. First, there is no evidence
of a bimodal distribution of plasma drug levels in populations
of northern European extraction. Second, sertraline has linear
pharmacokinetics even up to doses of 200 mg/day in terms of:
- No change in half-life from single dose to multiple dose
- No change in half-life over its full dosing range
- Proportional changes in plasma levels of both sertraline
and desmethylsertraline with dose increases
- No change in the ratio of sertraline to desmethylsertraline288
These findings are not consistent with the conversion of
sertraline to desmethylsertraline and its eventual elimination's
being substantially dependent on CYP 2D6 since sertraline
at a dosage of 150 mg/day does produce approximately a 50%
to 65% increase in the plasma levels of the CYP 2D6 substrate,
desipramine.153,298
Also, an immediate switch from fluoxetine to sertraline exerts
only a modest effect on sertraline and desmethylsertraline
levels under conditions that produce a 400% increase in the
levels of the desipramine. Third, the in vitro conversion
of sertraline to desmethylsertraline correlates more with
CYP 3A3/4 activity (r = 0.93) than with CYP 2D6 activity.209
Therefore, higher and lower doses, respectively, of sertraline
may be necessary when it is used in combination with drugs
that induce (eg, carbamazepine and phenytoin) and inhibit
(eg, ketoconazole) CYP 3A3/4.
The CYP enzymes responsible for the subsequent biotransformation
of desmethylsertraline have not been well characterized. Several
observations suggest that there is more than one potential
pathway for the further biotransformation of sertraline prior
to its elimination and that these pathways may be mediated
by more than 1 CYP enzyme. Several different metabolites have
been characterized in the plasma and/or urine of individuals
receiving sertraline. Additionally, sertraline is excreted
approximately equally in the feces and urine, suggesting that
there is more than one metabolite.
Metabolites
There has been considerable discussion about whether the
different SSRIs have "active" metabolites. One problem
is that these discussions often do not
begin with a definition of what is meant by the term "active"
and how such activity was assessed. The two primary metabolites
of fluvoxamine reportedly are not capable of inhibiting the
serotonin uptake pump,274
but no studies have been done on the effect of its numerous
metabolites on specific CYP activity.
Fluoxetine has a metabolite that is as potent and more selective
than the parent drug in terms of the inhibition of the serotonin
uptake pump (Table 3.8).
Since this metabolite has an unusually extended half-life
(ie, 7 to 15 days), its level and hence indirect serotonin
agonistic effects take time to fully develop and then persist
for an extended interval after the fluoxetine is discontinued.
Norfluoxetine is also more active than fluoxetine as an inhibitor
of CYP 3A3/4 and equal to fluoxetine as an inhibitor of CYP
2D6 (Table 8.7). Plasma
levels of norfluoxetine correlate with the magnitude and duration
of the inhibition of both of these enzymes following fluoxetine
administration.112,219
As with fluvoxamine, there are apparently no paroxetine
metabolites capable of inhibiting the serotonin uptake pump,
but the M2 metabolite is a potent inhibitor of CYP 2D6 (Table
8.7). There is no data on what plasma levels of this metabolite
could be expected in the normal population or any of the special
populations discussed later in this section.
Desmethylsertraline is 1/10th to 1/25th as potent as sertraline
at inhibiting the serotonin uptake pump (Table
3.8). Since its concentrations are only 1.5 times higher
than the parent drug under clinically relevant dosing conditions,219
it would be predicted to contribute only 6% to 15% (ie, 1.5
times higher levels times 1/10th to 1/25th the potency) to
the serotonin uptake inhibitory effects that would occur in
patients on sertraline under clinically relevant dosing conditions.
The magnitude of this contribution is probably too small to
be clinically meaningful in most situations. However, desmethylsertaline
like the major metabolites of fluoxetine
and paroxetine is virtually equipotent to the parent SSRI
as an inhibitor of specific CYP enzymes (Table
8.7). Hence, it would be expected to contribute to the
magnitude of such an effect. Since its half-life (62 to 104
hours) is longer than that of the parent drug, it would also
be expected to prolong the duration of the effect, but not
to the extent that norfluoxetine does.288
The clinical impact of this fact is mitigated by the relatively
weak inhibitory effect of sertraline and desmethylsertraline
on specific CYP enzymes.
Linear Versus Nonlinear Pharmacokinetics
Citalopram and sertraline show linear pharmacokinetics (ie,
changes in drug concentration proportional to the change in
dose). In contrast, fluvoxamine, fluoxetine and paroxetine
have nonlinear pharmacokinetics (Tables 6.4
and 6.5). The evidence for nonlinearity
with fluvoxamine and fluoxetine primarily comes from the observation
that their half-lives are substantially longer after multiple
dose administration than after single dose administration
(Table 6.4). The same observation also
holds for paroxetine, but in addition, there is good evidence
that under steady-state conditions,
the half-life of paroxetine is progressively longer at higher
doses.141
| TABLE 6.4 Change
in Half-life (t1/2) as a Function of Multiple Dose Administration |
| SSRI |
Single-
dose t1/2 |
Multiple-
dose t1/2 |
%
Change |
| Citalopram1 |
33 hours |
33 hours |
|
| Fluoxetine2
|
1.9 days |
5.7 days* |
 300
300 |
| Fluvoxamine3 |
15 hours |
22 hours |
 50
50 |
| Paroxetine4 |
10 hours |
21 hours |
200 |
| Sertraline5 |
26 hours |
26 hours |
|
* Only the effect
on t1/2 of fluoxetine is published.
Paroxetine has the shortest half-life of any SSRI in
terms of single dose which may increase the risk of withdrawal
reactions on this drug. Tapering the drug rather than
abrupt discontinuation should minimize such a reaction. |
| References: 1146,
223, 3234,
4141, 5288 |
The nonlinearity of paroxetine is also apparent based on
the fact that paroxetine plasma levels increase disproportionately
with the dose increases (Table 6.5).
In contrast, there is a linear relationship between dose and
plasma drug level increases with both citalopram and sertraline
(Table 6.5). Although fluoxetine has
been the most extensively-used SSRI and fluvoxamine has been
the longest-used SSRI, there are minimal data on what plasma
drug levels can be reasonably expected at different doses
administered long enough to achieve steady-state. Undoubtedly,
the long half-life of fluoxetine has discouraged such studies.
It would require almost 1 year to determine the steady-state
plasma levels of fluoxetine and norfluoxetine that could be
achieved in the same individual on the 4 different doses that
comprise its clinically relevant dosing range (ie, 20 to 80
mg/day).
Nonlinearity has the potential to be clinically significant
with these SSRIs for several reasons. While fixed-dose studies
with these drugs suggest that generally higher doses do not
produce a greater antidepressant effect, physicians frequently
try higher doses when the response has been suboptimal. Due
to nonlinearity, the concentration-dependent effects of fluvoxamine,
fluoxetine and paroxetine will be expected to increase disproportionally
with higher doses; that will not be expected with citalopram
or sertraline. This knowledge can be helpful to the clinician
in terms of what to expect with higher than usual doses of
these different SSRIs. The dose-dependent (ie, concentration-dependent)
effects
of the SSRIs are reviewed in Section
5.
| TABLE
6.5 Effects of Dose and Age on the Plasma Levels of
SSRIs |
| SSRI
|
Age
(yrs) |
Dose
(mg/day) |
Dose
Effect* |
Age
Effect |
| Plasma
Level (ng/ml) |
| Citalopram |
|
| 5 |
20 |
25 |
50 |
| 12 |
47 |
58 |
120 |
| NA |
109 |
NA |
NA |
|
|
| <
651 |
NC |
|
| > 652 |
|
133% |
| Fluoxetine
|
|
| 20 |
40 |
60 |
| 200 |
NA |
NA |
| NA |
NA |
NA |
|
|
| < 653 |
? |
|
| >
653 |
|
? |
| Fluvoxamine |
|
| 50 |
100 |
200 |
| NA |
93 |
250 |
| NA |
62 |
NA |
|
|
| < 654 |
25% |
|
| > 655 |
|
 33%
33% |
| Paroxetine |
|
| 20 |
30 |
40 |
| 49 |
86 |
129 |
| 79 |
147 |
228 |
|
|
| > 656 |
25% |
|
| > 656 |
|
50%
to 90% |
| Sertraline |
|
| 50 |
100 |
200 |
| 15§ |
29§ |
68§ |
| NA |
NA |
90 |
|
|
| <
657,8 |
NC |
|
| > 658 |
|
37%§ |
| NA = Not available;
NC = No change |
* (Plasma level on
dose [x + n] divided by plasma level on dose X) divided
by (dose [X + n] divided by dose X) minus 1, or D change
in plasma level, D change in dose minus 1 expressed as
%.
(Plasma level for the elderly group divided by plasma
level for the younger age group) minus 1 expressed as
%.
This result was extrapolated from the other values based
on the linearity of citalopram plasma levels in this age
group over this dose range.
§ These results are for males only. These results overestimate
the effect of age since young males develop lower plasma
levels on sertraline than do young females or older males
or females. The values for young females at 20 mg/day
is 104 ng/ml, which is essentially the same as for older
males and females. |
| References: 131,
295, 3Table
3.7, 473,
590, 6141,
7223, 8243 |
Half-life
Considerable variability among the SSRIs exists with regard
to half-life (Table 6.2). The half-life
of fluvoxamine is 15 to 22 hours.73
For this reason, and to reduce the incidence and severity
of nausea, it is generally administered in equally divided
doses twice a day. The half-lives of citalopram, paroxetine
and sertraline allow them to be administered once a day (Table
6.2). Fluoxetine and its active metabolite, norfluoxetine,
have unusually extended half-lives for orally administered
drugs: 2 to 4 days for fluoxetine and 7 to 15 days for norfluoxetine
(Table 6.2). Because of the extended
half-lives, this drug can be administered as infrequently
as once a week and still reach stable steady-state levels.
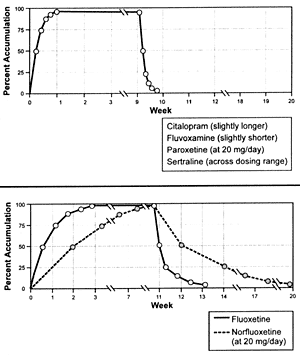
Figure 6.1 provides a graphic illustration
of the difference in duration of drug administration needed
to reach steady-state and the time to 95% washout following
drug discontinuation for fluoxetine and norfluoxetine versus
the other SSRIs. Due to this extended period, the magnitude
of any concentration-dependent effect of fluoxetine will take
several weeks to be achieved and will persist for several
weeks after it has been discontinued. The clinical consequences
may be either desirable or undesirable depending on the specific
situation. Such an extended half-life might provide an added
measure of safety against possible relapse if the patient
were intermittently noncompliant. While this proposal has
theoretical appeal, it was not substantiated by the relapse
prevention studies reviewed in Section 5 (Table
5.3). Instead, these studies show remarkably similar results
for fluoxetine, paroxetine and sertraline versus a parallel
placebo control. For the same theoretical reason, a slower
onset of antidepressant efficacy may be expected with fluoxetine
versus the other SSRIs, but there is no convincing data to
support that proposal. While data are lacking on the above
points, there are compelling data that the full magnitude
of fluoxetine-induced inhibition of CYP 2D6 and 3A3/4 is not
achieved until steady-state has been reached, and that full
recovery of enzyme activity is not achieved until fluoxetine
and norfluoxetine have fully washed out of the body.112,219
These points are clinically important because the gradual
accumulation and washout can affect the concentration of a
concomitantly prescribed drug administered to a patient taking
fluoxetine or recently discontinued from fluoxetine therapy.
The extended half-life also is responsible for the long interval
of washout that is recommended before initiating treatment
with a monoamine oxidase inhibitor (MAOI) following fluoxetine
discontinuation. At present, there is no good explanation
for the unusually long half-life of norfluoxetine.
The half-life of paroxetine is a function of its plasma
drug level. Following a single 20 mg dose, paroxetine has
a half-life of 10 hours (Table 6.4).
Paroxetine does not reach a half-life of 20 hours until steady-state
has been reached on 20 mg/day (Table 6.4)
because paroxetine inhibits CYP 2D6, which is the CYP enzyme
responsible for its biotransformation. When paroxetine levels
fall after this drug is discontinued, its clearance rate increases
as the inhibition of the enzyme is decreased. Thus, paroxetine
and fluvoxamine are
more quickly cleared from the body than the other SSRIs.
| FIGURE 6.1 Time
to Steady-state and Time to 95% Washout |
 |
|
Reference: 217
|
Rapid clearance may explain the increased incidence of withdrawal
reactions seen after these 2 SSRIs are discontinued in comparison
to the other SSRIs. Certainly, such withdrawal syndrome would
be highly unlikely with fluoxetine due to its extended half-life.
If such a reaction were to occur after abrupt SSRI discontinuation,
reinstituting the drug and more gradual tapering will commonly
handle the problem. This approach may be taken prophylactically
with fluvoxamine and paroxetine, particularly if the patient
previously had problems with abrupt discontinuation or if
the patient has been on higher than the usually effective,
minimum dose.
Effect of Age and Gender on Clearance
Although the reasons have not been elucidated, there is considerable
difference among the SSRIs with regard to changes in apparent
clearance of specific SSRIs in the physically healthy "young
old" (ie, in these studies, most of the "old"
individuals were between 65 and 75 years of age) versus younger
healthy individuals (Table 6.5). Plasma
levels of citalopram and paroxetine are approximately 100%
higher in the elderly compared with the young.95,141
Fluvoxamine has no apparent change in its metabolism as a
function of age;73,234,285
however, recent data suggest males develop plasma levels 40%
to 50% lower than females with the magnitude of the effect
possibly being greater at lower doses.118
The basis for this gender effect has not been established.
Fluoxetine has not been adequately studied, but data from
clinical trials suggest that plasma levels of fluoxetine plus
norfluoxetine can be twice as high in the elderly compared
to the young.86,159
There is an age by gender interaction for sertraline with
its plasma levels being 35% to 40% higher in elderly females
versus young males; however, sertraline plasma levels do not
differ between elderly females and elderly males or young
females.288
There are at least two reasons why these age related changes
in plasma drug levels can be clinically important:
- First, it will be predicted to increase treatment limiting
adverse effects in the elderly since the discontinuation
rate for SSRIs due to adverse effects is dose-dependent
and, hence, concentration dependent.
- Second, the inhibition of specific CYP enzymes by specific
SSRIs is concentration dependent; hence,
the elderly will be expected to experience a greater degree
of inhibition on average than younger patients given the
same dose of the same SSRI. The apparent order of the age
effect from most to least appears to be citalopram >
paroxetine ³ fluoxetine (probably, although not well
studied) > sertraline ³ fluvoxamine (Table
6.5).
The latter issue is important since the elderly are more
likely to be on concomitant therapy and more sensitive to
any adverse consequences produced by elevation of the levels
of such coadministered drugs. This phenomenon is particularly
relevant to fluoxetine and fluvoxamine due to the number of
CYP enzymes inhibited by these 2 SSRIs, which will be reviewed
in Sections 7 and 8.
Effect of Specific Organ Impairment
on Clearance
As discussed above, all of the SSRIs are dependent on oxidative
metabolism for their elimination, and the resultant polar
metabolites are primarily excreted via the urine. Based on
these facts, significant impairment in liver, renal and cardiac
function will be expected to affect the levels of either the
parent drug and/or its metabolites for each of the SSRIs.
Appreciable impairment in liver function and/or size can
slow the individual's ability to biotransform drugs and thus
result in greater drug accumulation per dose prescribed. Reduced
renal function generally leads to the increased accumulation
of polar metabolites that may be pharmacologically active,
either in a way similar to or different from the parent drug.
The accumulation of those metabolites can appreciably alter
the patient's response to the medication, including the risk
of adverse effects. Significant reduction in
left ventricular function will cause a reduction in hepatic
and renal arterial blood flow, which is another important
determinant of both hepatic- and renal-mediated clearance
of drugs. For these reasons, impairment in the function of
these organs can significantly alter a patient's response
to what is a usually therapeutic dose of a medication. The
effect of specific organ impairment on the clearance of specific
SSRIs has been studied to varying degrees with the different
SSRIs.
Based on single-dose studies, the half-lives of all the
SSRIs are approximately doubled in individuals with cirrhosis
compared to physically healthy individuals (Table
6.6). Since the clearance of fluoxetine and paroxetine,
to a substantial degree, and fluvoxamine, to a more modest
degree, is prolonged when going from a single dose to multiple
doses (Table 6.4), these single-dose
studies in patients with cirrhosis are likely an underestimate
of the magnitude of the effect of such impairment on the clearance
of these SSRIs. As with the elderly, the elevated levels of
specific SSRIs will
translate into greater inhibition of specific CYP enzymes
in this medically ill population.
| TABLE
6.6 Effect of Liver Disease on SSRI Metabolism
and Pharmacokinetics |
| Product |
Average
Half-life |
Healthy
Volunteers* |
Cirrhosis
Patients |
| Citalopram1 |
1.5 days |
3.5 day |
| Fluoxetine2 |
2 days |
7 days |
| Fluvoxamine3 |
15 hours |
24 hours |
| Norfluoxetine2 |
7 days |
12 days |
| Paroxetine4 |
12 hours |
20 hours |
| Sertraline5 |
1 day |
2 days |
|
* It must be remembered that
interindividual variability is high and that half-lives
of fluoxetine, fluvoxamine and paroxetine are
usually higher after multiple doses than after
a single dose.
|
| References:
113,2246,
3234,
468, 5288 |
|
| TABLE
6.7 Effect of Renal Impairment on Pharmacokinetics
of SSRIs (Single Dose) |
| SSRI |
Renal Impairment |
| Citalopram |
Not available |
| Fluvoxamine |
No effect1 |
| Fluoxetine |
No effect2 |
| Sertraline |
No
effect3 |
| Paroxetine |
100-150% in
plasma levels with GFR < 30 ml/min4 |
| References:
1234,
211, 3281,
478 |
|
The data on the effect of renal disease (Table
6.7) are even more limited than with cirrhosis particularly
because the effect should principally be an increase in plasma
levels of more polar metabolites, but the studies that have
been done have focused only on the parent drug. In a single-dose
study of paroxetine (30 mg) in individuals with renal impairment,
the plasma AUC and Cmax were significantly increased.281
Since the polar paroxetine metabolite, M2, is a potent inhibitor
of CYP 2D6, accumulation of this metabolite may be relevant
to the increase in paroxetine plasma levels in renally impaired
individuals and also in the elderly since renal function decreases
with age. The single-dose pharmacokinetics of fluvoxamine,234
fluoxetine,111 and
sertraline281,288
are similar in individuals with renal failure versus in healthy
volunteers. As discussed above, single-dose studies with fluoxetine,
and to a lesser extent with fluvoxamine, should be cautiously
interpreted since the nonlinear pharmacokinetics of these
drugs observed in healthy individuals (Tables 6.4
and 6.5) may well be increased in individuals
with such organ impairment.
There have been no studies of the effect of significant
left ventricular pump impairment on the clearance of any SSRI.
In the absence of data, it would be prudent to use conservative
dosing in such patients for the same reasons as discussed
with hepatic and renal impairment.
The disease-related changes in the clearance rates of the
SSRIs have several clinically important implications. Patients
with such organ impairment will need lower doses to achieve
the same plasma drug levels that occur in healthy individuals
on the usually effective, minimum dose. Hence, a dose reduction
will be appropriate for such individuals to compensate for
the reduction in their clearance; otherwise, they will be
at increased risk for having more of the dose-dependent adverse
effects of the SSRIs and a poorer antidepressant responses.
Additionally, these patients will be at increased risk for
pharmacokinetic drug-drug interactions. Since inhibition of
specific CYP enzymes produced by specific SSRIs is concentration-dependent
(see Section 8), the higher levels
that will occur in these patients without an appropriate dose
adjustment will result in a greater degree of enzyme inhibition.
The hepatic and cardiac impairment may also make these patients
more sensitive to the enzymes inhibiting effects of these
drugs (ie, a greater degree of enzyme inhibition may occur
in these individuals than those with normal organs at the
same concentration of the SSRI). There are no data on this
matter because the formal pharmacokinetic drug interaction
studies with the SSRIs have been done in
healthy individuals with normal organ function.
These issues are important because these patients, due to
their comorbid medical illnesses, are likely to be on multiple
medications and at greater risk for a drug-drug interaction.
Moreover, their medical illnesses may make them more susceptible
to any adverse effects arising out of such an interaction.
Being aware of these considerations can help the physician
make a prudent choice with regard to SSRI selection and dose
adjustment for a specific patient based on whether and to
what degree one or more organ(s) is impaired, what other medications
the patient is taking, and the overall health status of the
patient.
|



