|
|
| The Adverse Effect Profiles of the Selective Serotonin Reuptake Inhibitors: Relationship to In Vitro Pharmacology |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, May 2000, 153-157
|
In the preceding five columns,1-5 I have discussed the relationship between the in vitro binding affinity and the clinical pharmacology of the various classes of antidepressants. In this column, I will review this issue as it relates to the adverse effect profiles of the five marketed selective serotonin reuptake inhibitors (SSRIs).
Figure 1 was first presented and discussed in the second column in this series, "De-Spinning In Vitro Data," in which I discussed the clinical relevance of the in vitro pharmacology of the SSRIs.2 This figure shows the relative binding affinity of the different SSRIs for specific, clinically meaningful neural targets (i.e., uptake pumps, neuroreceptors). As can be readily seen, each of these drugs has at least one order of magnitude separation between its binding affinity for the serotonin uptake pump and its affinity for its next most potent potential site of action. A general rule in pharmacology is that a separation of at least one order of magnitude is needed for a drug to be considered "selective." In other words, a concentration of the drug can be achieved at which the drug will have a meaningful effect on its most potent site of action, while having no or only a minimal effect on its next site of action. Thus, Figure 1 captures the essence of the logic behind the word "selective" in the name of this class of antidepressants.
The clinically important question is whether the clinical pharmacology of the SSRIs is consistent with this concept of "selectivity." Certainly, they are all antidepressants. Moreover, all the SSRIs have approximately the same relative superiority over placebo in terms of both acute efficacy and relapse prevention based on double-blind, randomized clinical trials (Table 1).7-10
Another way to compare the clinical pharmacology of the SSRIs is to examine their relative risk of causing specific adverse effects. To make this comparison, I examined the incidence of adverse effects on each SSRI in comparison to the parallel placebo condition in the double-blind clinical trials used for the approval of these drugs by the Food and Drug Administration. Based on these data, I calculated the attributable incidence rate of adverse effects for each SSRI by taking the incidence of a given adverse effect in the group treated with the SSRI minus the incidence of the same adverse effect in the parallel group treated with placebo (Table 2). The attributed incidence is the incidence that is specifically due to the drug as opposed to being due to other factors such as the underlying illness (e.g., major depression). The attributed risk then is a clinically useful measure of how many patients are likely to experience the adverse effect when taking the drug compared to an identical group of patients not exposed to the drug.
By comparing across the columns in Table 2, the reader can estimate the likelihood of a specific adverse effect on one SSRI versus another, realizing that it is based on data from different studies as opposed to data from a single study. It would be preferable to have data from one large double-blind study in which patients were randomly assigned to either placebo or one of the five SSRIs at a comparable antidepressant dose. However, no such study has been done nor is ever likely to be done. There have been double-blind studies that directly compared the efficacy and tolerability of two SSRIs (i.e., head-to-head comparison studies). However, these studies frequently do not have a placebo control so that the attributable rate for each SSRI cannot be calculated. Given the differences that exist between clinical trials and clinical practice in terms of methodology and patient selection, the data shown in Table 2 may not reflect the actual rate of these adverse effects in a given practice.
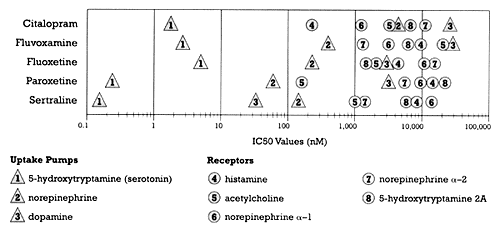 | Figure 1 - Relative Potency for Different Sites of Action for the SSRI Class of Antidepressants
- Based on data from Hyttel 1993 |
The same results are also shown in Table 3, perhaps in a more user-friendly way. In this table, I show the attributable incidence of given adverse effects infrequency categories such as > 7.5% but < 10%. Using this table, the reader can at a glance determine the relative likelihood of a specific adverse effect on a given SSRI relative to the other members of this class.
From my perspective, the data in both Tables 2 and 3 are completely consistent with the concept of the SSRIs as "selective." Virtually all of the adverse effects produced by one SSRI are produced by all SSRIs. Moreover, these adverse effects are consistent with the excessive agonism of serotonin produced by the SSRI-induced blockade of the neural uptake pump for serotonin. If these drugs affected another target to a pharmacologically significant degree, then I would expect that action to be reflected in the drug's adverse effect profile. As shown by Tables 2 and 3, that is not the case.
The differences that do appear in Tables 2 and 3 are quantitative rather than qualitative (i.e., the frequency rather than the nature of the adverse effect). For example, all SSRIs cause nausea but this adverse effect occurs most frequently on fluvoxamine. This adverse effect is likely mediated by excessive serotonin (5-hydroxytrytophan, 5-HT) agonism at the 5-HT3 receptor. The reason nausea occurs more frequently on fluvoxamine than on the other SSRIs is likely due to the pharmacokinetics of fluvoxamine. This SSRI is administered twice a day and at a higher total milligram dose in contrast to the other SSRIs. The frequency of this adverse effect appears to be dependent on what dose the stomach "sees" and how frequently the drug is administered, particularly early in treatment.14 Tolerance develops to this adverse effect in a time course that parallels down-regulation of the 5-HT3 receptor.14
| Table 1. SSRI versus placebo: Response rate and relapse rate | | Response Rate* (%)7 | Relapse Rate (%)8-10 | | SSRI | Placebo | Difference | P Value | Duration† | Placebo | SSRI | Difference | P Value | | Fluoxetine | 60 | 33 | 27 | < 10-13 | 52 | 57 | 26 | 31 | <0.01 | | Fluvoxamine | 67 | 42 | 25 | < 10-2 | NA | NA | NA | NA | NA | | Paroxetine | 65 | 36 | 29 | < 10-14 | 52 | 43 | 16 | 27 | < 0.01 | | Sertraline | 79 | 48 | 31 | < 10-11 | 44 | 46 | 13 | 33 | < 0.001 | | The meta-analysis for response rate did not include citalopram. A relapse prevention study has been done with catalopram, but it lasted 24 weeks rather than 1 year. A relapse prevention study has not been published for fluvoxamine.
*Response defined as at least 50% decrease in depression symptom severity, as measured using a standard instrument.
†Duration of maintenance treatment in weeks. |
To put these data in perspective, it is useful to review how adverse effects are assessed in clinical trials. Several approaches are actually used. One is to note any adverse event that occurs in the patient during the trial, whether or not the event is believed to be related to the treatment (e.g., the occurrence of dry mouth versus the occurrence of an automobile accident). The former may not be related to the investigational treatment (i.e., may occur at the same rate in patients receiving placebo as on active treatment), whereas the latter may be related to the investigational treatment (e.g., impairment in alertness, reaction time, or coordination). At the conclusion of the study, all reports of treatment-emergent adverse events are tallied and summarized by major categories (e.g., complaints of "dizziness," "light-headedness," "vertigo," "woozy" sensation, or "unsteadiness" may be all collapsed into the category "dizziness"). The incidence of a given adverse event category can then be compared across treatments in the same double-blind controlled study. The incidence of adverse effects in patients taking the investigational drug can be compared with the incidence in patients receiving placebo as one measure of evaluating whether the adverse event is drug related. The incidence can also be compared with that in patients taking active controls (i.e., agents marketed for the same indication) to determine whether the investigational drug has an advantage or disadvantage over an existing agent with regard to its adverse effect profile.
| Table 2. Comparison of the placebo-subtracted incidence rate (%) of frequent adverse effects for SSRIs* | | Adverse Effect | Citalopram11
(n=1063, 446)a | Fluoxetine12
(n=1730, 799)a | Fluvoxamine13
(n=222, 192)a | Paroxetine12
(n=421, 421)a | Sertraline12
(n=861, 853)a | | Anorexia | 2.0 | | 7.2 | | 8.6 | | 4.5 | | 1.2 | | | Confusionb | NA | | 1.5 | | NA | | 1.0 | | 0.8 | | | Constipation | < placebo | | 1.2 | | 11.2 | | 5.2 | | 2.1 | | | Diarrheac | 3.0 | | 5.3 | | -0.4 | | 4.0 | | 8.4 | | | Dizzinessd | < placebo | | 4.0 | | 1.3 | | 7.8 | | 5.0 | | | Drowsinesse | 8.0 | | 5.9 | | 17.2 | | 14.3 | | 7.5 | | | Dry mouth | 6.0 | | 3.5 | | 1.8 | | 6.0 | | 7.0 | | | Dyspepsia | 1.0 | | 2.1 | | 3.2 | | 0.9 | | 3.2 | | | Fatiguef | 2.0 | | 5.6 | | 6.2 | | 10.3 | | 2.5 | | | Flatulence | NA | | 0.5 | | NA | | 2.3 | | 0.8 | | | Frequent micturition | NA | | 1.6 | | 0.6 | | 2.4 | | 0.8 | | | Heache | < placebo | | 4.8 | | 2.9 | | 0.3 | | 1.3 | | | Increased appetiteg | NA | | NA | | NA | | NA | | NA | | | Insomnia | 1.0 | | 6.7 | | 4.0 | | 7.1 | | 7.6 | | | Nauseah | 8.0 | | 11.0 | | 25.6 | | 16.4 | | 14.3 | | | Nervousnessi | 3.0 | | 10.3 | | 7.6 | | 4.9 | | 4.4 | | | Palpitationsj | < placebo | | -0.1 | | NA | | 1.5 | | 1.9 | | | Paresthesiak | NA | | -0.3 | | NA | | 2.1 | | 1.3 | | | Rashl | NA | | 0.9 | | NA | | 1.0 | | 0.6 | | | Respiratorym | 8.0 | | 5.8 | | -1.3 | | 0.8 | | 0.8 | | | Sweating | 2.0 | | 4.6 | | -1.3 | | 8.8 | | 5.5 | | | Tremors | 2.0 | | 5.5 | | 6.1 | | 6.4 | | 8.0 | | | Urinary retentionn | < placebo | | NA | | NA | | 2.7 | | 0.9 | | | Vision disturbances | < placebo | | 1.0 | | 0.0 | | 2.2 | | 2.1 | | | Weight gaing | NA | | NA | | NA | | NA | | NA | | | *The adverse effect data presented above come from product labeling as opposed to head-to-head trials. Such data may not necessarily reflect the actual rate of these adverse effects in clinical practice or the actual differences between these various drugs.
aThe first value is the number of patients on that medication, while the second represents those treated in the parallel placebo group.
bIncludes decreased concentration
cIncludes gastroenteritis
dIncludes lightheadedness, postural hypotension, and hypotension
eIncludes somnolence, sedation and drugged feeling
fIncludes asthenia, myasthenia and psychomotor retardation
gReported incidence < 1% in both drug-treated and parallel placebo-treated groups. Included in this table to facilitate comparison with adverse effects tables for other medications in subsequent columns
hIncludes vomiting
iIncludes anxiety, agitation, hostility, akathisia and central nervous system stimulation
jIncludes tachycardia and arrhythmias
kIncludes sensation disturbances and hypesthesia
lIncludes pruritis
mIncludes respiratory disorder, upper respiratory infection, flu, dyspnea, pharyngitis, sinus congestion, oropharnyx disorder, fever and chill.
nIncludes micturition disorder, difficulty with micturition and urinary hesitancy. |
| Table 3. The most likely adverse effects on specific SSRIs above and beyond the parallel placebo condition (percentage on drug minus percentage on placebo based on registration studies)* | | SSRI | > 7.5% | > 10% | > 15% | > 20% | > 25%† | > 30% | | Citalopram | drowsiness
respiratory
nausea | -- | -- | -- | -- | -- | | Fluoxetine | -- | nausea
nervousness | -- | -- | -- | -- | | Fluvoxamine | anorexia
nervousness | constipation | drowsiness | -- | nausea | -- | | Paroxetine | dizziness
sweating | fatigue
drowsiness | nausea | -- | -- | -- | | Sertraline | insomnia
diarrhea
tremors | nausea | -- | -- | -- | -- | | * Data in this table are based on the information presented in Table 2. The adverse effect data presented above come from product labeling as opposed to head-to-head trials. Such data may not necessarily reflect the actual rate of these adverse effects in clinical practice or the actual differences between these various drugs.
† Best available data suggest that all SSRIs can cause sexual dysfunction (e.g., delayed ejaculation, decreased libido, anorgasmia) in approximately 30% of patients. |
Several limitations need to be considered when analyzing these data (Table 4). First, the incidence rate for a specific adverse effect may be dose-dependent. Ideally, one would prefer to compare the incidence rates for doses that are comparably effective; however, this is rarely done for several reasons. There may be controversy or uncertainty about what are equivalently effective doses. Many clinical trials are done using a flexible rather than a fixed dose design so that the largest database for each drug represents the incidence rate over the entire dosing range used in the development program.
Second, the incidence rates do not distinguish whether the adverse event occurred once during treatment (e.g., a "cold"), was an intermittent problem (e.g., headaches), or was persistent (e.g., anorgasmia or dry mouth). Of course, the same limitation is also true for the placebo condition, so that the drug versus placebo difference should be meaningful.
| Table 4. Limitations of the incidence rate of adverse effects as assessed in clinical trials | - May be dose-dependent
- Dose not indicate whether the event is a one-time, intermittent, or persistent occurence
- Does not reflect severity
|
Third, the incidence rate does not reflect severity. One drug may have an appreciably lower rate of adverse effects than another. However, that apparent advantage may be offset by the fact that the adverse effects are either so intolerable or so serious that treatment must be discontinued. One way of addressing this issue is to look at the discontinuation rates for different drugs due to adverse effects.
All of these caveats should be kept in mind when evaluating the data presented in this column. In the next column, I will review similar adverse effect data for the non-SSRI antidepressants and consider how their adverse effect profiles are related to their affinities for various neural mechanisms of action.
References
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Preskorn SH, De-spinning in vitro data. J Pract Psychiatry Behav Health 1999;5:283-7
- Preskorn SH, Two in one: The venlafaxine story. J Pract Psychiatry Behav Health 1999;5:346-50
- Preskorn SH, Bupropion: What mechanism of action? Journal of Psychiatric Practice 2000;6:39-44
- Preskorn SH, Imipramine, Mirtazapine, and Nefazodone: Multiple targets. Journal of Psychiatric Practice 2000;6:97-102
- Hyttel J, Comparative pharmacology of selective serotonin reuptake inhibitors (SSRIs). Nord J Psychiatry. 1993;47 (suppl 30):5-12
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Montgomery SA, Dufour H, Brion S, et al, The prophylactic efficacy of fluoxetine in unipolar depression. Br J Psychiatry. 1988;153(suppl 3):69-76
- Doogan DP, Caillard V, Sertraline in the prevention of depression. Br J Psychiatry. 1992;160:217-222
- Eric LT, A prospective double-blind comparative multicentre study of paroxetine and placebo in preventing recurrent major depression episodes. Biol Psychiatry. 1991; 29(suppl 11):2545
- Physicians’ Desk Reference, 54th Edition. Prescribing information for citalopram. Montvale, NJ: Medical Economics Co.; 2000:1073-7
- Preskorn SH, Comparison of the tolerability of bupropion, fluoxetine, imipramine, nefazodone, paroxetine, sertraline and venlafaxine. J Clin Psychiatry. 1995;56(suppl 6):12-21
- Compendium of Pharmaceuticals and Specialities. 33rd edition. Ottawa: Canadian Pharmaceutical Association; 1998:922-24
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
|



