|
|
| Two in One: The Venlafaxine Story |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, November 1999, 1-5
|
In this series of columns, I have focused on how in vitro binding affinity of drugs is related to their clinical effects. In my last column,1 I discussed why the differential binding affinity of selective serotonin reuptake inhibitors (SSRIs) for different neural sites of action allows these drugs to selectively affect only the serotonin uptake pump at the concentrations usually achieved at their recommended dosing range. In this column, I will use venlafaxine to illustrate a drug with a binding affinity that sequentially affects two different neural sites of action over its clinically relevant dosing range. I will specifically address the following questions:
- Why does the adverse effect profile of venlafaxine qualitatively change as its dose is increased, in contrast to the adverse effect profile of an SSRI?
- Why does venlafaxine have an ascending dose-antidepressant response curve in contrast to the flat doseantidepressant response curve seen with SSRIs?
- Why are higher concentrations of venlafaxine needed to achieve antidepressant efficacy in comparison to several of the SSRIs?
Figures 1a and 1b show the binding affinity of the various SSRIs and venlafaxine, respectively, in terms of clinically important neural mechanisms of action. Please note that Figures 1a and 1b are based on the results of different in vitro studies.
As I have explained in earlier columns, binding affinity is important because it tells you what a drug is capable of doing. That fact is captured in the following equation:
(Equation 1)
Effect = affinity for x drug x biological
site of action level variance
Binding affinity does not tell you what a drug will do but rather how much drug is needed to produce a specific effect -- that is, what drug level is needed to engage a site of action to a physiologically meaningful degree. This is because binding affinity or potency is a concentration-dependent term. Drug level in turn dictates the dosing rate that is needed in relation to the patient's ability to clear the drug. This relationship is expressed in the following equation:
(Equation 2)
Concentration = Dosing rate / Clearance
Relative binding affinity
Figures 1a and 1b can be used in two different ways. First, they can be used to compare the affinity of one drug for different targets. This permits one to estimate the difference in dose that is needed to achieve concentrations that will produce effects mediated by these different targets. Second, these figures can be used to compare the relative ability of different drugs to affect different targets. This information in turn can be used to estimate the concentrations that are needed for two different drugs to engage the same target to a physiologically meaningful degree. I will use venlafaxine as an example to illustrate both of these uses.
The dose-dependent adverse effect profile of venlafaxine
Of all the sites shown in Figure 1b, venlafaxine most avidly binds to the serotonin uptake pump. It is five times more potent (i.e., has a higher binding affinity) as an inhibitor of the serotonin versus the norepinephrine uptake pump.3 That difference in binding affinity is the same as the difference between its lowest recommended dose for treating major depression, 75 mg/day, and its highest, 375 mg/day. As discussed in the first column in this series ("Defining Is"), this 5-fold gap in binding affinities raises the possibility that venlafaxine sequentially inhibits serotonin and norepinephrine uptake pumps over its clinically relevant dosing range. Such a sequential action would be consistent with venlafaxine's dose-dependent adverse effect profile and its ascending dose-antidepressant efficacy curve.7
The adverse effect profile of venlafaxine at a dose of 75 mg/day is comparable to that of an SSRI, which is consistent with the fact that its highest binding affinity is for the serotonin uptake pump.7 Its adverse effect profile suggests that a dose of 75 mg/day of venlafaxine produces concentrations that only meaningfully inhibit the serotonin uptake pump. At higher doses, the adverse effect profile of venlafaxine, in contrast to an SSRI, also begins to include effects consistent with norepinephrine potentiation.7 For example, the risk of increased blood pressure is clearly a dose-dependent effect. At doses under 100 mg/day, the incidence of sustained elevation of supine diastolic blood pressure in patients on venlafaxine is comparable to that found in patients on placebo.8 However, the incidence is three-fold greater than on placebo in patients taking more than 200 mg/day of venlafaxine.8 This observation suggests that, at doses around 200 mg/day, venlafaxine begins to block the norepinephrine uptake pump sufficiently to produce this effect.
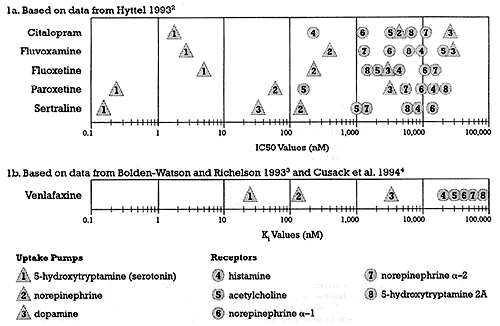 | Figure 1 - Relative Potency for Different Sites of Action for five SSRIs and Venlafaxine
Note that the data in Figure 1a are expressed in terms of 50% inhibition concentration, whereas the data in Figure 1b are expressed as kinetic inhibition constants. For these reason, the data cannot be directly compared across the two figures Instead, the goal of these figures is to show the relative gap in potencies of different drugs for inhibiting different targets
The report by Hyttel was used to generate the data shown in Figure 1a because it had data for all five marketed SSRIs; however, it did not contain data on venlafaxine.2 The in vitro studies used to generate the data shown in Figure 1b provided data on venlafaxine but not on all five SSRIs.3,4 While proofing this column, I found a recent in vitro study that included data on all six drugs.5 The data from this later study are the source of the kinetic inhibition constants shown in Table 1. Of note, this study found a greater difference in the potency of venlafaxine for inhibiting the serotonin versus norepinephrine uptake pumps than had previously been reported. This study used cloned human cells, whereas earlier studies used synaptosome preparations. While the use of cloned human cells should be an advantage, the potency gap reported in this study is not consistent with the clinical pharmacology of venlafaxine. I raise this issue here to underscore the point that significant differences in the results of in vitro data do occur and must be taken into account when trying to extrapolate from such results to the clinical situation. l will discuss this issue at greater length in a subsequent column. |
The ascending dose-antidepressant response curve of venlafaxine
The ascending dose-antidepressant response curve seen with venlafaxine is also consistent with this theory of the sequential inhibition of uptake pumps. Drugs that selectively block either the serotonin or the norepinephrine uptake pump (e.g., the SSRIs and desipramine, respectively) have antidepressant properties -- suggesting that either of these mechanisms of action are capable of mediating an antidepressant response. Furthermore, clinical trial data indicate that some patients respond to one mechanism of action but not the other.9 This suggests that there may well be different biochemical forms of depression that are responsive to these different mechanisms of action. Finally, the combination of an SSRI and a norepinephrine selective reuptake inhibitor (NSRI) (e.g., maprotiline or desipramine) has been reported to produce a faster and greater antidepressant response than a SSRI alone, suggesting that these two mechanisms of action can be synergistic in terms of mediating antidepressant efficacy.10 These observations, along with the sequential engagement of the serotonin and then norepinephrine uptake pumps, fit with the fact that venlafaxine has an ascending dose-antidepressant response in contrast to the flat dose-antidepressant response curve seen with the SSRIs.7 This putative dual mechanism of action is also consistent with the finding that treatment with high doses of venlafaxine produced a greater antidepressant response than treatment with fluoxetine, 40 mg/day, in a double-blind study of hospitalized patients with melancholia.11 In fact, this dual mechanism of action theory for venlafaxine served as the rationale for a recently completed study in which the relative efficacy of venlafaxine versus citalopram was tested in patients who had historically not benefited from previous SSRI treatment (for a discussion of such a study design, see my July 1997 column12). The results of that study, when fully analyzed, will hopefully provide empirical evidence about the relative merits of switching among the SSRIs in the case of nonresponse versus switching to an antidepressant with a different or additional mechanism of action.
We tested this theory about sequential inhibition of the serotonin and then the norepinephrine uptake pumps in man using the biochemical measures for each and found the following:13 Venlafaxine at both 75 mg/day and 375 mg/day produced a substantial degree of serotonin uptake inhibition in platelets as did sertraline 50 mg/day. In contrast, the NSRI, maprotiline, at 150 mg/day had no effect on these measures. Conversely, venlafaxine, 375 mg/day, and maprotiline, 150 mg/day, blocked the tyramine pressor effect (a norepinephrine-mediated response), whereas venlafaxine, 75 mg/day, and sertraine, 50 mg/day, did not. These results indicate that venlafaxine at a dose of 75 mg/day achieves a concentration sufficient to block the serotonin but not the norepinephrine uptake pump, whereas, at 375 mg/day, venlafaxine achieves a concentration sufficient to block both of these mechanisms of action. These results provide a heuristic explanation that ties together the in vitro binding affinity of venlafaxine for the serotonin and norepinephrine uptake pumps and its dose-dependent clinical pharmacology with respect to both efficacy and tolerability.
How does this information on venlafaxine's binding affinity fit with the plasma levels needed for different antidepressants to be therapeutically effective?
| Table 1. Relationship between dose, usual drug level, plasma drug level (bound and unbound), kinetic inhibition rate constant, and magnitude of serotonin uptake inhibition in platelets for the SRIs shown in Figure 1.* | | | | | | Plasma drug levels | | | Magnitude of | | SRI | Dose
(mg) | Usual
drug level
(ng/ml) | Percent
protein
bound | Free drug
level
(ng/mL) | Kinetic
inhibition
rate constantb | SE uptake
inhibition
in platelets | | citalopram | 40 | | 85 | | 50% | | 40 | | 1.16 | | 60% | | | fluoxetine | 20 | | 200a | | 95% | | 10 | | 0.83 | | 80% | | | fluvoxamine | 200 | | 250 | | 75% | | 60 | | 2.22 | | 80% | | | paroxetine | 20 | | 40 | | 95% | | 2 | | 0.13 | | 80% | | | sertraline | 50 | | 25 | | 95% | | 1 | | 0.29 | | 80% | | | venlafaxine | 75 | | 105a | | 25% | | 75 | | 9.09 | | 80%c | | | * Table modified from Preskorn 199614 and 199915
a Combined concentration of parent drug and nearly equipotent, active metabolite
b Kinetic inhibition constant (nM) from Leonard and Richelson5
c From Harvey and Preskorn 1997.13 At a dose of 375 mg/day, the average concentration of venlafaxine and desmethylvenlafaxine is 528 ng/ml and the magnitude of serotonin uptake inhibition is 95%. |
Historically, psychopharmacologists have been enamored by in vitro binding affinity and relatively disinterested in plasma drug levels. However, the point of this series of columns is that in vitro binding affinity is only relevant from the perspective of drug concentration. Table 1 shows the kinetic inhibition rate constant, the plasma drug level (bound and unbound), and the degree of serotonin uptake inhibition produced by each of the five-marketed SSRIs and venlafaxine. Several hypotheses/conclusions can be drawn from this table. First, antidepressant response to an SRI requires that the patient be on a dose that produces a concentration at which the drug will inhibit the serotonin uptake pump by 60%-80%. Second, there is, in general, good agreement between the drug's binding affinity for the serotonin uptake pump and the concentration of the drug needed to produce 60%-80% serotonin uptake inhibition.
The major caveat to this last statement is that the total venlafaxine plasma levels in Table 1 may seem lower than expected relative to the drug's binding affinity. The likely explanation is a difference in protein binding. The drug level shown in Table 1 is total plasma drug level (i.e., bound plus unbound). The unbound portion is what is in equilibrium with the uptake pump. Venlafaxine has a much lower protein binding (25%-30%) than do the SSRIs (80%-100%). Thus, the free fraction of venlafaxine is much higher than the other drugs at the same total plasma drug level.
It should be noted that each of these drugs at the doses shown in Table 1 has about the same antidepressant efficacy in terms of either response or remission rates based on either double-blind, placebo-controlled or double-blind, head-to head comparisons.7 These results strongly suggest that these drugs work through serotonin uptake inhibition to produce this antidepressant response. The difference is that results of double-blind, fixed-dose studies show that venlafaxine has an ascending dose-antidepressant response curve whereas the SSRIs do not.7 This is consistent with the fact that, at increasing doses, venlafaxine engages another mechanism of action -- norepinephrine uptake inhibition -- that is also capable of mediating antidepressant response. In contrast, the SSRIs do not do this because of the larger gap between their binding affinity for the serotonin uptake pump and their next potential neural mechanism of action (Figure 1).
Conclusion
In this column, I have expanded on the theme of this series -- the relationship between in vitro binding affinity and clinical pharmacology. Here I have used venlafaxine to illustrate the differences between an antidepressant with two mechanisms of action over its clinically relevant dosing range and the SSRIs with their single mechanism of action. The pharmacology of venlafaxine provides an illustration of how critical drug concentration is to understanding the relationship between binding affinity and clinical effects.
The goal of this column was not to take a position on whether a single action or a dual action drug is better but rather to explain the pharmacological principles underlying each approach. With that knowledge, clinicians can judge for themselves the relative merits of either approach. After all, the upside and downside are flip sides of the same pharmacology.
On the upside, a medication with dual action has a built-in augmentation strategy that may be helpful for a specific patient. On the downside, the patient cannot experience the benefits of the dual action without also being exposed to its potential drawbacks. In the case of a high dose of venlafaxine, this is the risk of increased blood pressure. There is also a risk of two types of pharmacodynamic drug interactions. For example, as a result of venlafaxine's effects on both serotonin and norepinephrine uptake, the patient who is taking a high dose of venlafaxine is at risk for both the serotonin syndrome and a hypertensive crisis, when venlafaxine is used in combination with monoamine oxidase inhibitors.
Another downside is that a dual-action drug is in essence a fixed combination product. For example, venlafaxine is essentially a fixed combination equivalent to 4 parts SSRI for every 1 part of NSRI. Hence, the prescriber cannot vary the degree of action on one mechanism (e.g., serotonin uptake) without also affecting the degree of action on the other mechanism (e.g., norepinephrine uptake). For that reason, the patient on venlafaxine must always experience more serotonin than norepinephrine potentiation.
In contrast, a physician could use a SSRI and a NSRI in combination and, by varying their doses individually, would be able to vary the relative effect on each mechanism independent of the other. However, this strategy has its downsides. First, it is likely to be more expensive, particularly if neither drug is available in a generic version. Second, it may be more complicated for the patient to follow and thus may increase the chances that the patient may inadvertently not take the medications as prescribed.
For a more extended discussion of these issues, the reader is referred to my recent book Outpatient Management of Depression: A Guide for the Primary-Care Practitioner. In the end, practitioners will make their choices based on their assessment of the relative upside and downside of each approach and their knowledge of the circumstances surrounding their patient. Parenthetically, the same principles discussed here are applicable to the area of antipsychotics where we have drugs with a single mechanism of action (e.g., haloperidol) as well as drugs with dual or even multiple mechanisms of action (e.g., risperidone and olanzapine, respectively).
In my next column, I will extend this discussion to a consideration of the clinical relevance of the in vitro binding affinity of another antidepressant with a dual mechanism of action, bupropion.
References
- Preskorn SH, De-spinning in vitro data. J Pract Psychiatry Behav Health 1999;5:283-7
- Hyttel J, Comparative pharmacology of selective serotonin reuptake inhibitors (SSRIs). Nord J Psychiatry. 1993;47 (suppl 30):5-12
- Bolden-Watson C, Richelson E, Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029
- Cusack B, Nelson A, Richelson E, Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559-565
- Leonard BE, Richelson E, Synaptic effects of antidepressants: Relation to their therapeutic and adverse effects. In: Buckley PF, Waddington JL, eds. Schizophrenia and mood disorders: The new drug therapies in clinical practice. Oxford: Butterworth Heinemann; 1999: [AU: PAGE NUMBE
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Physicians’ Desk Reference, 53rd edition. Montvale, NJ: Medical Economics Company, 1999
- Thase ME, Rush AJ, When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(suppl 13):23-29
- Nelson JC, Mazure CM, Bowers MB Jr, Jatlow PI, A preliminary, open study of the combination of fluoxetine and desipramine for rapid treatment of major depression. Arch Gen Psychiatry. 1991;48:303-307
- Clerc GE, Ruimy P, Verdeau-Pallés J, A double-blind comparison of venlafaxine and fluoxetine in patients hospitalized for major depression and melancholia. The Venlafaxine French Inpatient Study Group. Int Clin Psychopharmacol. 1994;9:139-143
- Preskorn SH, Appearance of knowledge. J Pract Psychiatry Behav Health 1997;3:233-8
- Harvey AT, Preskorn SH, Mechanism of action of venlafaxine in normal male volunteers (Abstract). Clin Pharmacol Ther 1997;61:175
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Preskorn SH, Baird S, 11.2 billion dollars: Is it worth it?. J Pract Psychiatry Behav Health 1998;4:163-8
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
|



