|
|
| Polypharmacy in a Patient with Refractory Major Depression: Part I: The Case |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice Vol 8, No. 6, November 2002, 370-376
|
This column is another in the clinical pharmacological case series. The goal of this series is to illustrate basic principles relevant to the daily practice of clinical psychopharmacology. Just as for other cases in this series, the following two equations are relevant to our understanding of the case presented here:
(Equation 1)
Effect = affinity for x drug x biological variance
site of action level
(Equation 2)
Drug concentration = closing rate / clearance
The case will be presented and discussed in two parts. In this first column (Part I), the case is presented followed by a series of questions for the reader to consider. The first and most immediate questions are then discussed. Part II will continue the discussion with a consideration of the remaining questions and the implications of the case for clinical management.
This case dates back to the era when the tricyclic antidepressants (TCAs) were the mainstay of treatment for major depression. Nevertheless, the principles it illustrates are also relevant to the use of newer psychotropic agents, such as the selective serotonin reuptake inhibitors (SSRIs), other newer antidepressant agents, and the atypical antipsychotics.
Now and at the time of this case, the dose that a patient receives is generally determined using a dose titration approach based on clinical assessment of response,1 as illustrated in Figure 1.
| Figure 1 - Dose titration based on clinical assessment of response | 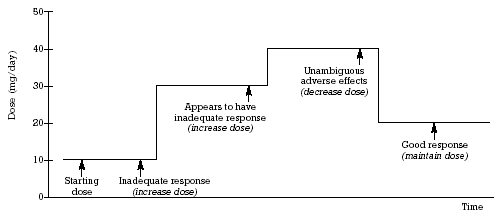 |
Briefly, this approach involves beginning with a dose that is either the usual starting dose or the usually effective dose, depending on whether the drug requires titration to reach the usually effective dose. For example, tertiary amine tricyclic antidepressants (TCAs), such as amitriptyline, are drugs that generally require titration to an effective antidepressant dose, whereas the SSRI antidepressants can usually be started at an effective antidepressant dose and generally do not require titration. Following a reasonable period of time, the clinician evaluates the patient’s response; if it is inadequate and the patient is not experiencing adverse effects, the dose is increased until the desired response is achieved. If adverse effects occur, the dose is either reduced or the patient is switched to a different antidepressant. This was the standard approach when tertiary amine TCAs were the antidepressants of choice and is still the most common method of dose titration used in practice when titration is necessary. (For additional information on this topic, see the section "The Rationale for and Clinical Implications of Dose-Response Curves")
A 35-year-old male with treatment-refractory major depression
The patient was initially seen for clinical depression by his primary care physician, who started the patient on amitriptyline. When the patient had not benefited from a trial of 150 mg/day, his primary care physician referred him to a psychiatrist who admitted him to a psychiatric hospital. The physical examination on admission revealed tachycardia, tremulousness, and mild ataxia. The patient’s blood pressure was normal, but was measured only at the time of admission, which was the common practice on psychiatric units then and remains so today. The patient’s medical history was unremarkable and there was no history of substance abuse or dependence. The admission diagnosis was major depression, marked severity.
The electrocardiogram (EKG) on admission confirmed sinus tachycardia; there were no other EKG abnormalities. No laboratory abnormalities were found in the workup, which included normal complete blood count, normal electrolytes, normal protein and albumin, normal liver function tests, and normal urinalysis with a negative urine screen for drugs of abuse.
The patient denied any problems with anticholinergic side effects such as dry mouth, constipation, or urinary retention. Given that fact and the inadequate response, the amitriptyline dose was increased to 250 mg/day.
After 8 days, the patient complained of generalized anxiety, a panic episode, recurrent bouts of palpitations, and headaches. The psychiatrist concluded that the depressive episode was worsening with the development of more prominent anxiety symptoms. Given this assessment and no complaints of anticholinergic side effects, the amitriptyline dose was increased to 300 mg/day and the following medications were added: propranolol, 20 mg three times a day, diazepam, 5 mg four times a day, and flurazepam, 30 mg at night.
After 2 weeks, the patient was more listless, disinterested and confused. The assessment was worsening of the depressive episode. Given this assessment and no complaints of anticholinergic side effects, the amitriptyline dose was increased to 350 mg/day. Three days later, the patient complained of intermittent chest pain and swelling of his feet. Furosemide (Lasix) was added to treat these complaints.
Questions: What would you do next in this case? What do you think happened next?
Continuation of case report
The next day this patient was found in his hospital bed unresponsive, without respiration or pulse. An autopsy was performed 5 hours later and revealed no anatomical cause of death on either gross or microscopic inspection. Specifically, there were no pill fragments in the stomach, no cardiomegaly, no cirrhosis, no pulmonary disease, no pulmonary emboli, and no brain pathology. The total tricyclic antidepressant blood level postmortem was 5540 ng/ml (2600 ng/ml of the parent drug, amitriptyline and 2940 ng/ml of the primary metabolite, nortriptyline). The sample for this analysis was obtained from the inferior vena cava.
Questions
This case raises numerous questions, listed below in reverse chronological order:
- Why did this patient die?
- Why did this patient develop swelling of his feet?
- Why was this patient likely hypotensive?
- Why were the postmortem levels of amitriptyline so high?
- Why did this patient become more listless, disinterested, and confused?
- Why did this patient develop increased anxiety symptoms?
- Why did this patient not respond to the initial trial of amitriptyline?
The answer to the first question about cause of death is obvious in one way, but not so obvious in another. For that reason, the second and third questions listed above will be addressed first to provide background information relevant to a more complete answer to Question 1.
Why did this patient develop swelling of his feet?
Some readers may wonder whether this patient developed an arrhthymia that accounted for the peripheral edema. However, cardiac causes of peripheral edema require right-sided pump failure. An arrhthymia is an unlikely cause and would not present as an otherwise asymptomatic peripheral edema.
As with any medical complaint, there is a differential diagnosis for bilateral peripheral edema. Unfortunately, nothing was done antemortem in this case to evaluate the cause. Therefore, the possible causes must be considered within the context of what is known in terms of the patient’s admission workup, which was rather complete, his course in the hospital, and his autopsy.
Venous insufficiency is a common cause of lower extremity edema. However, primary venous insufficiency is unlikely to develop over 3 weeks in an otherwise healthy, young adult. Peripheral edema could result secondary to venous thrombosis, but that is unlikely to be bilateral and otherwise asymptomatic (e.g., no calf pain or tenderness). In addition, there was no evidence of emboli at the time of the autopsy. However, the veins in the legs were not specifically inspected, so that this explanation cannot be completely ruled out.
Another cause of peripheral edema is hypoproteinemia, including hypoalbuminemia, which can occur as a result of malnutrition or glomerulonephropathy. There are several reasons to rule out these explanations. The patient was not malnourished on admission or at the time of his death. His serum protein and albumin levels were normal on admission. His urinalysis was normal, and his kidneys were normal both grossly and microscopically at the time of his death.
On the other hand, there is an explanation that is consistent with all of the available data: substantial alpha-1 adrenergic receptor blockade secondary to amitriptyline. While amitriptyline is classified as an antidepressant, it is actually polypharmacy in a single pill (i.e., it has multiple mechanisms of action over the concentration range that can be achieved under usual clinical dosing).2
The effects of amitriptyline as an alpha-1 adrenergic receptor blocker can perhaps be best understood by examining the effects of the five drugs classified as alpha-1 adrenergic blockers in the 2002 Physicians’ Desk Reference (PDR).3 These drugs are doxazosin (Cardura) (p. 2668-71), phenoxybenzamine (Dibenzyline) (p. 3457-9), terazosin (Hytrin) (p. 464-7), prazosin (Minipress) (p. 2699-2701), and the combination product, prazosin plus polythiazide (Minizide) (p. 2700-1). While most of these drugs were initially developed as antihypertensives, two of them (doxazosin and terazosin) are now also indicated for the treatment of benign prostatic hyperplasia.
The use of these agents as antihypertensives was limited by their propensity to cause marked lowering of blood pressure, especially orthostatic hypotension and syncope. The most potent action of these five drugs is their ability to block alpha-1 adrenergic receptors; as such, they can cause syncope after the first dose or within the first few days of therapy. That has led to a bolder warning in the package inserts for all five of these drugs (i.e, "class" labeling).
In contrast, alpha-1 adrenergic blockade is at least the sixth most potent effect of amitriptyline and occurs at about the same concentration as the antidepressant effect of the drug.2 The first mechanism of action of amitriptyline is histamine-1 receptor blockade, followed by muscaranic cholinergic receptor blockade. These receptor actions cause sedation and anticholinergic effects, respectively, although the severity can vary among patients. It is because of these multiple mechanisms of action, that amitriptyline has to be titrated. This is also the reason why orthostatic hypotension is less common, less severe, and presents later in the course of treatment with this drug than with the five classic alpha-1 adrenergic receptor blockers mentioned above. A graphic illustration of the multiple concentration-effect relationships with amitriptyline was presented in an earlier column in this series.4
In fact, the package insert for most of the classic alpha-1 adrenergic receptor blockers recommends titration to minimize the risk of orthostatic hypotension and syncope. Some of these drugs are even produced in dosage strengths below the usually effective dose principally to make dose titration possible.
In addition to syncope, peripheral edema is an adverse effect listed in the package insert for all five of the classic alpha-1 adrenergic receptor blockers listed above and was up to four times more common in patients receiving these drugs in clinical trials than in patients in the parallel placebo groups. This peripheral edema is directly linked to the physiology underlying the drugs’ blood pressure lowering effects. By blocking alpha-1 adrenergic receptors, these drugs reduce the muscular tone on precapillary arterioles and thus reduce peripheral vascular resistance. As a result, the capillary vasculature volume increases and can cause peripheral edema.
Parenthetically, the package inserts for these classic alpha-1 adrenergic blockers illustrate how much pharmacologic knowledge and drug development have progressed between the time when the earlier alpha-1 adrenergic blockers, such as phenoxybenzamine, were developed and the introduction of later alpha-1 adrenergic blockers such as terazosin. The package insert for phenoxybenzamine is less than one page long and has minimal information on the pharmacodynamics and pharmacokinetics of the drug, and hence on drug-drug interactions, and minimal systematic information concerning the incidence of adverse effects. In contrast, the package insert for terazosin is three and a half pages long and has considerably more information on these matters. Returning to the case, recall that the physician added the loop diuretic furosemide (Lasix) in response to the development of peripheral edema. This addition to the treatment regimen was important, as will be discussed below.
Why was this patient likely hypotensive?
Unfortunately for this discussion and more unfortunately for this patient, his blood pressure was only measured at the time he was admitted to the hospital. Therefore, it can only be conjectured that he was hypotensive later in his hospitalization; however, hypotension is consistent with the medications he was taking and with his clinical course. An explanation of this statement requires knowledge of both the pharmacological actions of the drugs the patient was taking and some basic equations from cardiovascular physiology:
Blood pressure = cardiac output x total peripheral (CO) resistance (TPR)
Cardiac output = heart rate x stroke volume (HR) (SV)
Thus, Blood pressure = HR x SV x TPR
As explained above, this patient was on a drug that can decrease TPR, the alpha-1 adrenergic receptor blocker, amitriptyline. It is this mechanism that accounts for orthostatic hypotensive episodes on amitriptyline, which can lead to falls and broken hips in vulnerable patients (e.g., elderly).
This patient was also receiving a drug that reduces CO by reducing both HR and SV, the beta adrenergic receptor blocker, propranolol. This is the mechanism for the negative chronotropic and ionotropic effects of propranolol on the heart and accounts for its antihypertensive effects.
The package inserts for most of the classic alpha-1 adrenergic blockers specifically warn--sometimes in bold, black letters--about the risk of hypotension in patients receiving an alpha-1 adrenergic receptor blocker "who are also receiving a beta blocker such as propranolol" (package insert for Minipress, PDR, p. 2700). Although the phenoxybenzamine insert is relatively short, it specifically warns that the combined use of alpha-1 and beta adrenergic receptor blockers can "produce an exaggerated hypotensive response" (PDR, p. 3458).
This patient was at risk for a pharmacodynamically mediated drug-drug interaction (DDI) in which one drug (propranolol) decreased CO while another drug (amitriptyline) decreased TPR.
The physician then added furosemide, a potent loop diuretic. Furosemide inhibits the absorption of sodium and chloride principally in the loop of Henle (hence the term "loop diuretic") as well as in the proximal and distal tubules. The most likely explanation for the addition of furosemide is that the physician thought the patient was fluid overloaded, another cause of edema. However, there was no indication in the chart that the patient was ingesting excessive amounts of fluid and his autopsy revealed no evidence of fluid overload.
Nevertheless, the addition of furosemide would be expected to cause a diuresis resulting in a decrease in the patient’s vascular volume on the day before he died. While the package insert for furosemide is not in the 2002 PDR, reflecting the age of the drug, it is in the 1994 PDR.5 There, the precaution section warns of the potential that "excessive diuresis may cause dehydration and blood volume reduction with circulatory collapse" 5 (p.1042).
The addition of furosemide thus set the stage for another pharmacodynamic DDI, which would further compromise this patient’s cardiovascular status. The reduction in vascular volume decreases preload on the heart, which should have further decreased the patient’s blood pressure following the Frank-Starling principle from basic cardiovascular physiology. Again, this explanation is plausible but only a conjecture, since the patient’s blood pressure was not monitored.
| Figure 2 - Schematic illustration of the three-way pharmacodynamic drug-drug interaction that likely led to the fatal outcome in this case | 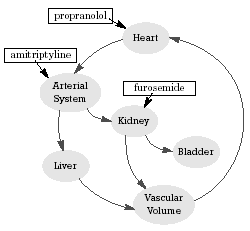 |
Thus, this case illustrates the potential for a triple pharmacodynamically mediated DDI in which each drug affected a different site of action but all of these sites of action were important to this patient’s cardiovascular system (Figure 2). The drugs and their sites of action were: propranolol (beta adrenergic receptors), amitriptyline (alpha-1 adrenergic receptors), and furosemide (Na+/K+/Cl- pump in the renal tubule, principally in the loop of Henle). Of note, the precaution section in the 2002 package insert for prazosin states that the "addition of a diuretic or other antihypertensive has been shown to cause an additive effect." The 2002 package insert for doxazosin also warns that "additional antihypertensives should be added with caution." The 1994 package insert for furosemide also warns about its ability to potentiate the effects of other antihypertensive drugs, including peripheral adrenergic blocking drugs, as well as about its potential to cause hypokalemia.
Why did this patient die?
The simple answer is that the patient died from an overdose of amitriptyline, given the nature of the death, the absence of another cause of death based on the autopsy findings, and the high TCA levels found in the postmortem blood. However, this simple answer does not address the physiological cause of death nor the question of whether the overdose was intentional (i.e., suicide) or unintentional (i.e., slow accumulation of a toxic concentration due to an excessive dosing rate relative to clearance). These issues are discussed below.
The reader may wonder whether the physiological mechanism underlying this patient’s death was cardiovascular collapse. This cannot be ruled out with absolutely certainty because the death was not observed. However, there is another more likely possibility.
Ventricular arrhythmia is a common mechanism of sudden death; it is also a common occurrence in TCA overdoses as discussed in previous columns.4,6 Given this possibility, it is relevant that this patient was on three drugs that can directly or indirectly delay various components of the intracardiac conduction system. Amitriptyline at high concentrations can block fast Na + channels.2 Propranolol can delay conduction through the atrial-ventricular (AV) node. Furosemide does not directly affect intracardiac conduction but can cause a loss of K +, which is why a potassium supplement is usually prescribed in conjunction with furosemide, although that was not done in this case. Hypokalemia can cause arrhthymias because the resting membrane potential of the myocyte is K + dependent. Thus, this patient was at risk for another three-way pharmacodynamic DDI adversely affecting his intracardiac conduction and setting the stage for a fatal arrhythmia.
In this case, amitriptyline was almost undoubtedly the critical agent in the fatal outcome. Ironically and unfortunately, excessive effects on the fast Na + channel can delay intracardiac conduction to the point that the patient is prone to develop ventricular arrhythmias, which can lead to sudden death. This explains the narrow therapeutic index of tricyclic antidepressants and their concentration-dependent lethality in drug overdoses, whether intentional or unintentional.
As noted in a previous column,2 tertiary amine tricyclic antidepressants such as amitriptyline can be considered multiple medications in a single pill with different actions predominating at different concentrations. The ability of tertiary amine tricyclic antidepressants to block fast Na + channels is the reason these drugs can be used as type I antiarrthymics. For example, imipramine, another tertiary amine tricyclic antidepressant like amitriptyline, was used for this purpose in the Cardiac Arrhythmia Pilot Study.7 For the interested reader, two previous columns have reviewed cases of unintentional overdoses of tricyclic antidepressants.4,6
Why were the postmortem levels of amitriptyline so high in this patient?
Such levels indicate an overdose--the question is whether it was an acute, intentional overdose on the part of the patient or a chronic, unintentional overdose. Before discussing this differential diagnosis, a note of caution must be given about interpreting postmortem levels of drugs such as amitriptyline, which have a large volume of distribution and are highly protein bound. As illustrated in the case titled "What Happened to Tommy?"4 , postmortem levels of such drugs are increased relative to antemortem levels. In that case, a plasma level was obtained within 30 minutes before death and then a postmortem blood level was obtained several hours after death. The postmortem level was approximately 5 times higher than the antemortem level. Why?
Recall that the postmortem sample in the case of the 35-year old man described here (and in most such cases) was whole blood, which was to some degree autolysized. In contrast, antemortem levels are based on plasma, which is prepared by spinning down a fresh whole blood sample to remove the red blood cells, white blood cells, and platelets. From a pharmacokinetic perspective, red blood cells, white blood cells, and platelets are a tissue compartment. Highly lipophilic and protein-bound drugs such as amitriptyline are found in higher concentration in tissue compartments than in plasma. Hence, levels in whole blood reflect both tissue concentration as well as plasma concentration and will thus be higher than the plasma concentration alone. In addition, there may be redistribution from other tissue compartments into the blood as the body decomposes. However, that mechanism was not likely to be a major factor in this case because the autopsy was performed relatively quickly after death. Taking into account the time between the patient’s death and the postmortem sample as well as the outcome of the case, the patient likely had a TCA plasma level well above the minimum toxic threshold of 450 ng/ml8 and above the seriously cardiotoxic level of 1,000 ng/ml.9,10
Two facts from the autopsy results make an acute intentional overdose unlikely. First, there were no pill fragments in the stomach, which is generally the case in an acute overdose as the patient typically dies during the absorption phase. Second, the nortriptyline level was slightly higher than the amitriptyline level in the postmortem sample, which is generally the case under steady state conditions in patients undergoing routine therapeutic drug monitoring. In deaths due to acute overdoses, the level of the parent drug is almost invariably much higher than the metabolite. This is because the patient overdoses only on the parent drug and dies during the absorption phase before the parent drug can be converted into the metabolite. Recall that nortriptyline is produced by demethlyation of amitriptyline, which is enzymatically mediated and requires functioning cytochrome P450 enzymes--and the patient must be alive for this biotransformation to occur. These two facts from the autopsy coupled with the patient’s clinical course lead to the conclusion beyond a reasonable doubt that this patient died of an unintentional chronic overdose.
Conclusion
Based on the preceding discussion, with the advantage of retrospection, it can be seen that each dosing decision in this case worsened the patient’s situation. Yet the clinician was clearly trying to achieve the best possible outcome for the patient.
Why was the outcome so untoward despite the best of intentions and the use of the dosing approach that is still the most commonly used in clinical practice, dose titration based on clinical assessment of response? The answer to this overarching question as well as the following subset of question will be the subject of part II of this case presentation:
- Why did this patient become more listless, disinterested, and confused?
- Why did this patient develop increased anxiety symptoms?
- Why did this patient not respond to the initial trial of amitriptyline?
- Why did the psychiatrist keep increasing the dose of amitriptyline?
- What could the psychiatrist have done differently?
As was pointed out at the beginning of this column, this case dates back to the time when the TCAs were the primary psychopharmacological tool for the treatment of major depression. Nevertheless, the principles it illustrates are still relevant to the safe and effective use of both older and newer medications whether alone or in combination. This case as well as earlier cases in this series,4,6 also illustrates why the TCAs have generally been replaced as first line agents for the treatment of major depression: They have multiple effects and a narrow therapeutic index (TI) that can cause serious toxicity at high concentrations. The goal of this column, as of all of the columns in this clinical psychopharmacology series, is to provide real life examples of such basic principles on the premise that much can be learned from such untoward outcomes that will help prevent future bad outcomes.
References
- Preskorn SH, Have you phenotyped your patient lately? J Pract Psychiatry Behav Health 1996;2(2):115-7
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Physicians’ Desk Reference, 56th Edition. Montvale, NJ:Medical Economics Company; 2002
- Preskorn SH, What happened to Tommy? J Pract Psychiatry Behav Health 1998;4:363-7
- Physicians’ Desk Reference, 48th Edition. Montvale, NJ:Medical Economics Company; 1994
- Preskorn SH, Clinical pharmacology case conference: A suicide attempt? Journal of Psychiatric Practice 2002;8:306-10
- Bigger JT Jr, Methodology for clinical trials with antiarrhythmic drugs to prevent cardiac death: US experience. Cardiology 1987;74(Suppl 2):40-56
- Preskorn SH, Jerkovich GS, Central nervous system toxicity of tricyclic antidepressants: phenomenology, course, risk factors and the role of therapeutic drug monitoring. J Clin Psychopharmacol. 1990;10:88-95
- Petit JM, Spiker DG, Ruwitch JF, et al, Tricyclic antidepressant plasma levels and adverse effects after overdose. Clin Pharmacol Ther 1977;21:47-51
- Preskorn SH, Fast GA, Therapeutic drug monitoring for antidepressants: Efficacy, safety and cost effectiveness. J Clin Psychiatry. 1991;52(suppl 6):23-33
Suggested Readings
- Baunwald E, Fauci A, Kasper DL, et al. Harrison’s principles of internal medicine, 15th edition. New York: McGraw-Hill; 2001
- Guyton AC, Hall JE. Textbook of medical physiology, 10th edition. Philadelphia: WB Saunders; 2000.
- Hardman JG, Limbird LE. Goodman & Gilman’s the pharmacological basis of therapeutics, 10th edition. New York: McGraw-Hill; 2001.
|



