|
|
| Polypharmacy in a Case of Refractory Major Depression: Part II: Implications for Clinical Management |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice Vol 9, No. 1, January 2003, 71-78
|
This column continues the discussion of the clinical pharmacology case started in the last issue.1 The brief summary that follows and the information presented in Figure 1 should allow the reader to follow the discussion in this column. Nevertheless, some may wish to read Part I in the previous issue of the journal.*
The Case
The patient was a 36-year-old man with a major depressive episode. He was initially treated with the tricyclic antidepressant (TCA), amitriptyline, by his primary care physician who titrated his dose from 50 to 150 mg/day over a period of a couple of weeks (Figure 1). However, he experienced no benefit and therefore was referred to a psychiatrist who hospitalized him consistent with the prevailing standard of care for refractory patients at the time of this case.
The admission workup revealed a physically healthy male suffering from a nonpsychotic but severe major depressive illness, with no comorbid psychiatric syndromes. Given the absence of response and the absence of appreciable anticholinergic adverse effects, the psychiatrist increased the dose of amitriptyline gradually over the course of the next couple of weeks to 300 mg/day. During the course of this upward titration, the patient’s depressive syndrome appeared to worsen. First, more anxiety symptoms emerged, for which propranolol, diazepam, and flurazepam were added. The patient then grew more confused and lethargic, leading the psychiatrist to increase the amitripytline dose to 350 mg/day. The patient next developed swelling of his feet for which furosemide (Lasix) was added. One day later, he was found dead. A postmortem examination revealed no anatomical cause of death. The patient’s postmortem level of amitriptyline + nortriptyline was 5540 ng/ml.
In the last column, the following questions were addressed:
- Why did this patient die?
- Why did this patient develop swelling of his feet?
- Why was this patient likely hypotensive?
- Why were the postmortem levels of amitriptyline so high?
This column will address the following additional questions raised by this case:
- Why did this patient become more listless, disinterested, and confused?
- Why did this patient develop increased anxiety symptoms?
- Why did this patient not respond to the initial trial of amitriptyline?
- Why did the psychiatrist keep increasing the dose of amitriptyline?
- What could the psychiatrist have done differently?
Just as with earlier clinical pharmacology cases reviewed in this column, the following two equations are relevant to answering the questions listed above:
(Equation 1)
Effect = affinity for x drug x biological variance
site of action level
(Equation 2)
Drug concentration = closing rate / clearance
| Figure 1 - Time course of medications | 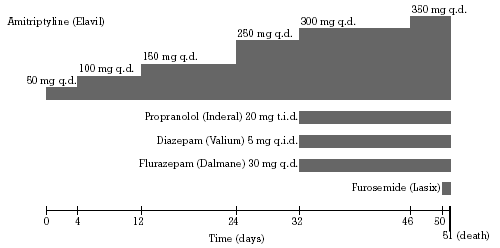 |
Why did this patient become more listless, disinterested, and confused?
The psychiatrist in this case interpreted these signs and symptoms as the result of worsening of the depressive illness and as evidence that the dose of amitriptyline was inadequate. Although this could have been the case, these symptoms could also have been occurred because of the medications the patient was receiving rather than in spite of them.
In terms of equation 1, the patient was taking three different medications (amitriptyline, diazepam, and flurazepam) that can all produce sedation because of their sites of action. The most potent effect of amitriptyline is histamine-1 receptor blockade.2 All tertiary amine TCAs block histamine-1 receptors, which is why they can be used at low doses as sedative and as antipruritic agents. In fact, doxepin is available in a topical preparation to treat various pruritic skin conditions. However, this patient appeared to be relatively insensitive to the histamine-1 blocking actions of amitriptyline based on the data in his medical records, at least until diazepam and flurazepam were added to his medication regimen.
Diazepam and flurazepam are both benzodiaepines and potentiate the effect of the inhibitory neurotransmitter, gamma-aminobutyric acid (GABA) by binding to the benzodiazepine binding site. GABA causes hyperpolarization of neurons by opening the chloride channels on these cells.3 In the presence of benzodiazepines, the concentration-response curve for the effect of GABA on the chloride channel is shifted to the left. A left shift is another way of saying its effect is potentiated or saying a given concentration of GABA produces a greater than normal effect. This is why benzodiazepines are anxiolytics and sedatives, and it is why they can potentiate the sedative effects of a variety of other medications including alcohol, barbiturates, and central histamine-1 blockers such as amitriptyline. Parenthetically, the latter is a pharmacodynamic drug-drug interaction (DDI) and was the likely cause of the patient’s increased listlessness, disinterest, and confusion. Unfortunately, these symptoms were attributed to a worsening of the patient’s depressive illness and hence led to a further increase in the dose of the one of the offending drugs (i.e., amitriptyline) (Figure 1).
According to its package insert, propranolol can also cause lethargy, lassitude, fatigue, and decreased performance on neuropsychometric tests as well as a depressive syndrome.4 The mechanism and physiology underlying these effects are less well established, but they may be mediated by excessive reduction in cardiac output or represent a central effect mediated by beta adrenergic receptor blockade. As discussed in Part I, this patient’s blood pressure was checked only once at the time of his admission to the hospital, so that there are no definitive data concering his blood pressure later in the course of treatment.
Why did this patient not respond to the initial trial of amitriptyline and why did the patient become anxious as the dose was increased?
There are several possible explanations. The physician in this case believed the dose was inadequate on the basis of the lack of response and no appreciable anticholinergic adverse effects. For that reason, he began the upward dose titration shown in Figure 1.
A second possibility is that the patient had a depressive illness that was not responsive to the mechanisms of action of amitriptyline, regardless of its dose.
A third possibility is that the 150 mg/day dose may ironically have been too high rather than too low. TCAs such as amitriptyline can cause dose-dependent neurotoxicity, specifically delirium and seizures. However, the earliest symptoms of TCA-induced neurotoxocity can mimic worsening of depressive and anxiety symptoms.5 The physical examination and EKG upon admission revealed tachycardia, tremulousness, and mild ataxia, which are all consistent with the early stages of amitriptyline intoxication. As happened with this patient, these symptoms can lead the prescriber to increase the dose of the TCA until the patient presents with unmistakable TCA intoxication: seizures, delirium, arrhthymias, or sudden death.5
How could this patient have been toxic on 150 mg/day of amitriptyline?
As explained in equation 2 above, the level of a drug is dependent on the dosing rate (i.e., the total daily in this case and most cases) relative to the patient’s ability to clear the dose. This patient was Caucasian and approximately 8% of Caucasians are genetically deficient in the human drug metabolizing enzyme, cytochrome P450 (CYP) 2D6.6 This enzyme mediates the ring hydroxylation of TCAs, which is usually the rate-limiting step in their clearance. Over 50 different mutations of this gene have been identified. Some code for a nonfunctioning CYP 2D6 enzyme, others for less efficient forms of the enzyme, and still others for more efficient forms of the enzyme.
Individuals deficient in CYP 2D6 have a substantially reduced ability to clear TCAs and other CYP 2D6 preferential substrates compared to the vast majority of humans. On the same dose, they will develop levels of CYP 2D6 substrate-drugs 3-4 times higher than CYP 2D6 normal metabolizers. For this reason, individuals deficient in CYP 2D6 have been termed CYP 2D6 poor metabolizers (PMs).
The supposition that this 36-year-old man was a CYP 2D6 PM fits parsimoniously with all of the facts of the case. This case thus illustrates the clinical relevance of genetically determined interindividual biological variance among patients (i.e., variable 3 in Equation 1) that can shift the dose-response curve for a given patient. In the instance of CYP 2D6 deficiency, the patient is more sensitive to the effects of a given dose of a CYP 2D6 substrate drug because the drug concentration produced by that dose is 3-4 times higher than what would normally be expected. In other words, it is as if the patient was taking a dose 3-4 times higher than the prescribed dose (equation 2).
How could the physician in this case have tested this possibility?
The basic goal of therapeutic drug monitoring (TDM) with any drug is to assess the appropriateness of the dose relative to the patient’s ability to clear the drug (equation 2). If one is certain of the dose (i.e., no compliance problems), then clearance can be determined by dividing the dosing rate by the drug level:
(revised Equation 2)
Clearance = dosing rate / drug concentration
In the case of this 36-year-old man, the dosing rate was known with considerable confidence since the patient was hospitalized and dosing was done under nursing supervision. Unfortunately, TDM was not done, although a postmortem TCA level was obtained and was consistent with a chronic overdose as explained in Part I of this column.
Was there any other reason this patient developed toxic levels of amitriptyline?
As discussed in Part I, this patient likely developed a significant decrease in blood pressure as amitriptyline accumulated in his system. This decrease in blood pressure was then further accelerated by the addition of propranolol. Together these two drugs decreased the patient’s cardiac output and his total peripheral resistance.
These decreases in turn led to greater accumulation of amitriptyline because cardiac output determines hepatic and renal arterial blood flow. The liver and the kidneys cannot clear a drug from the blood any faster than the blood is delivered to them. That is why patients with impaired cardiac output develop unusually high levels of TCAs even if they are genetically normal metabolizers of these drugs.7 This patient thus was caught in a vicious cycle in which higher levels of drug decreased his cardiac output, which in turn led to further increases in drug accumulation.
Why did the psychiatrist keep increasing the dose of amitriptyline?
At the time that this physician was treating this patient, there was a widely held belief that depressed patients were often underdosed with TCAs. For example, in an article published in the Journal of the American Medical Association in 1982, Keller et al. stated:
At least 150 mg/day of imipramine hydrochloride (or amtriptyline hydrochloride) or its equivalent is considered to be the average minimum effective dose. Doses of between 250 and 400 mg for at least four weeks are recommended for patients whose conditions do not improve.8
In treatment guidelines published that same year, Appleton stated:
No patient should be deemed refractory until tried on 300 mg per day [of imipramine or its equivalent] for four weeks. A few patients require 450 mg or more daily and can receive it safely.9
The scientific basis for the safety claim for doses of 450 mg or more daily in the Appleton guidelines is uncertain and does not apply to patients who are CYP 2D6 deficient or who have low clearance because of renal, hepatic, or cardiac disease. Nevertheless, the psychiatrist in this case was well within the dosing guidelines recommended in these two 1982 articles. The final dose in this case was in fact below the maximum recommended in both of these articles. The psychiatrist in this case was also following the standard dose titration approach of increasing the dose based on a clinical assessment of inadequate response and no apparent rate-limiting adverse effects. The problem with this approach is that the physician has to determine whether the inadequate response is because the dose is too low or too high. That can be difficult, especially if the patient does not report or demonstrate the adverse symptoms (in this case, peripheral anticholinergic effects) that the prescriber is expecting. That is as true today as it was at the time of this case and is a problem with a number of psychiatric medications besides TCAs. The admonitions to use higher doses of TCAs in these two and other articles of the day were based principally on anecdotal clinical experience that increasing the dose could convert some nonresponders into responders. There is in fact a genetic explanation for this recommendation -- but it holds for only a minority of patients who are genetically determined ultrarapid metabolizers (UMs) via CYP 2D6. The percentage of the population who are CYP 2D6 UMs varies from 1% or less in some Oriental populations10,11 to 3%-8% of Caucasians12 to 30% of some populations in North Africa and the Persian Gulf.10 Parenthetically, the percentage of CYP 2D6 PMs generally varies inversely with the percentage of CYP 2D6 UMs within a given population, perhaps reflecting differences in diets important to survival. In 20%-30% of cases, CYP 2D6 UM status is due to stable duplication or multiplication of the CYP 2D6 gene. In the remaining 70%-80% of cases, the reason for CYP 2D6 UM status is unknown but may be due to a genetic mutation producing a more efficient variant of the enzyme.
CYP 2D6 is only one human drug metabolizing enzyme but it illustrates the problem of finding one dose of a drug that will work for all individuals within a population and for all populations in the world when the drug is principally dependent on a single polymorphic enzyme such as CYP 2D6 for its clearance. This phenomenon poses problems for drug developers as well as prescribers,13 but that is a topic for another column in the series on the human genome project and drug development.14 It is sufficient for this discussion to use CYP 2D6 to point out the challenges posed by interindividual variability among patients (variable 3 in equation 1).
When, if at all, should TDM have been performed in this case?
TDM is an approach that specifically determines the ability of a given patient to clear a drug (equation 2) taking into account the mechanisms that are relevant to interindividual variance, including genetic status and the effects of intercurrent disease and concomitant medications. It can also be used to detect noncompliance, which is another major cause of less than optimal outcomes. Despite these facts, at the time of this case and also today, TDM of most psychiatric medications is relatively rarely employed in practice. The reader might consider when TDM might have been used in this case:
- Would it have been done prior to or at the time of referral to the psychiatrist?
- Would it have been done at the time of the apparent worsening of the illness despite the dose escalation (or in this case probably because of the dose escalation)?
- Would it have been done when the swelling of the feet was noted?
- Or would it not have been done until the time of autopsy?
This case has been presented to many clinical audiences, with the presentation stopped at the point when the patient’s feet began to swell and the audience asked what they would do. Only about one third of attendees have suggested doing TDM at that point. Thus, the fact that TDM was not done in this case would fit with the standard of care even today as defined by what the majority of practitioners would or would not do in this case.
The following summary statement made by Burrows and Norman in 1981 is relevant to this matter:
The question of the routine analysis of plasma concentrations of tricyclic antidepressants has caused considerable debate. Isolated plasma level determinations may aid, but cannot substitute for, sound clinical judgment. If an upper limit of tricyclic antidepressants does exist, then it is most likely determined by their toxic effects rather than by other pharmacological factors. (p. 197)15
It is of interest that the debate referenced by Burrows and Norman was ongoing in the early 1980s, despite several important facts that were well known at the time. First, overdoses of TCAs routinely produce serious and even fatal toxicity whether the overdose is intentional or unintentional, acute or chronic. Second, there is 40-fold variability in the clearance of TCAs among physically healthy individuals.16,17 Third, TDM can detect such variability and could guide appropriate dose adjustment to compensate for such differences using equation 2. In fact, as a result of their studies of twins, Alexanderson et al. determined in 1969 that this large interindividual variability in the clearance of TCAs was principally genetically mediated -- even though it would take two more decades for the responsible gene, CYP 2D6, to be isolated, and it was not until the past decade that the 50 plus mutations that account for the variability in metabolic status among individuals in diverse populations were discovered.18-20
In essence, this column covers almost 40 years of research discoveries, ranging from the first findings concerning the serious toxicity produced by substantial overdoses of TCAs in the early 1960s, to the large interindividual variability in TCA clearance found in the late 1960s, to the role of TDM in the safe prescribing of TCAs when given in antidepressant doses (i.e., 150 mg/day or more) which was identified between the early 1970s and late 1980s, and to the genetic basis for the variability in TCA clearance which was elucidated between the late 1980s and the present day.
What are the limitations of dose titration based on clinical assessment of response?
This approach to dose titration is based on the premise that well trained and experienced clinicians can identify mild and reversible adverse effects of the drug before serious and/or irreversible adverse effects can occur. Problems arise when it is difficult to distinguish between early signs of drug toxicity and symptomatic worsening of the patient’s psychiatric illness.
In cases such as this one, the untoward outcome is frequently cited as evidence that the prescriber did not use "sound clinical judgment." Restated, this position would be: "If the judgment had been sound, then there would not have been an untoward outcome." However, that is simply a tautology and no controlled study has ever been done to determine what percentage of well-trained and conscientious clinicians can detect the difference between symptoms due to an underlying psychiatric illness and symptoms due to the drugs used to treat them. The literature is replete with case examples in which clinicians appear in retrospect to have misinterpreted adverse drug effects as a worsening of the patient’s neuropsychiatric condition. Some examples have been reviewed in case studies published in earlier columns.21-23 In 9 of the 36 cases (i.e., 25%) of TCA-induced neurotoxcitiy (delirium and seizures) reviewed by Preskorn and Jerkovich, the clinician misinterpreted the early signs of neurotoxicity as a worsening of depression and increased the dose, just as appears to have happened in this case.5
While the case reviewed here dates back a number of years, the problem of proper attribution continues to be relevant in psychiatric practice and can even be made more difficult when polypharmacy is involved because of the potential for complex drug-drug interactions. It may be more difficult with some newer drugs than it is with the TCAs, because the TCAs produce numerous peripheral adverse effects that can usually be easily distinguished from adverse behavioral effects. In this case, the physician was focusing on peripheral anticholinergic adverse effects to determine whether the dose was too high. He specifically noted throughout the chart the absence of peripheral anticholinergic adverse effects. The absence of those effects coupled with a lack of efficacy formed the basis for the decision to increase the dose. This is the logic behind the concept of dose adjustment based on clinical assessment of response (Figure 1 in Part I of this column1), which was the common practice of the day when this case occurred and is still the most common method of adjusting the dose of most psychiatric medications.
The problem is that some patients -- such as the one in this report -- are for unknown reasons relatively immune to the peripheral anticholinergic effects of TCAs. The patient described in this case was not alone in this regard. Two previous columns have also described patients who either died or nearly died as a result of chronic TCA poisoning without developing noticeable peripheral anticholinergic adverse effects.24,25 The same was true for 36 cases of TCA-induced neurotoxicity reviewed by Preskorn and Jerkovich.5 In fact, absence of peripheral anticholinergic adverse effects is a risk factor for developing TCA-induced delirium, because clinicians make dosing decisions based on the presence or absence of such peripheral anticholinergic symptoms just as the clinician in this case did. Such resistance to peripheral TCA-induced anticholinergic adverse effects may perhaps represent another genetically determined difference among patients but that is a topic for another column.
Are there lessons from this case that are relevant to today’s practitioner?
While amitriptyline is an old drug, it is still widely used. Thus, many of the points are directly applicable to patients who are receiving this medication today, even though high doses such as those used in this case are probably rarely used today. Beyond patients receiving amitriptyline or another TCA, there are a number of basic principles that can be abstracted from this case and applied to the prescription of more recently developed drugs:
1. The adverse effects of psychiatric medications can present as worsening of the psychiatric illness being treated. As mentioned above, this principle is even more important with newer psychiatric medications because they have been developed as much as possible to affect only the brain. To the degree this goal is realized, the adverse effects of these medications will affect the brain and thus will frequently present as psychiatric symptoms rather than as peripheral adverse effects (e.g., dry mouth or constipation).
2. Some drugs are in essence multiple drugs in a single pill and hence the effects that predominate will be a function of the drug concentration achieved in the patient. While amitriptyline is clearly such a drug, so too are many of the newer antipsychotics.26 Whereas antidepressants have moved from drugs with multiple mechanisms of action, such as the TCAs, to newer agents with a single mechanism of action, such as the SSRIs, the opposite has been true with antipsychotics where the movement has been from single mechanism of action drugs such as haloperidol to multiple mechanisms of action drugs such as the various atypical antipsychotics. Those multiple mechanisms of action are believed to account for the ability of these newer antipsychotics to treat deficit as well as positive symptoms in patients with schizophrenia and also for their lower propensity to cause both acute and chronic extrapyramidal side effects. While such a profile may be beneficial, this pharmacologic characteristic also means that the effects of these drugs can vary substantially depending on the concentration of the drugs achieved in a given patient.
3. TDM can help the clinician determine whether a given dose is appropriate for a specific patient. There are few laboratory tests available to help diagnose psychiatric disorders or guide treatment decisions due in large measure to the lack of knowledge about the pathophysiology of most psychiatric disorders. As discussed above, TDM is one laboratory approach that can objectively assess the appropriateness of a dose of drug relative to the patient’s ability to clear that drug. From this perspective, TDM is simply a refinement of the approach of dose titration based on clinical assessment of response. In dose titration approach, the clinician via his or her examination of the patient is the assay methodology. The clinician is in essence assessing (or assaying) the patient’s response (the product in equation 1) to answer the following question: "Have I engaged the right site of action (variable 1 in equation 1) (by the virtue of the drug I have recommended to the patient) to the right degree (variable 2) (by virtue of the dose I have recommended) in the right patient (variable 3)?"
The highly subjective nature of response assessment in psychiatric disorders is the principal limitation of this dose titration approach. Clinical assessment of response is a process dependent on the clinician’s interpretation of what the patient reports (i.e., content) and how they report it (i.e., process). With TDM, if the dosing rate is known with confidence (i.e., no compliance issues) and the drug concentration is measured, then clearance can be mathematically calculated (revised Equation 2). That value can then be compared to the "usual" clearance in the "usual" patient enrolled in the clinical trials that led to the approval of the drug. Actually, a slight variation is most often done in practice. In this variation, the clinician does not calculate the clearance but instead compares the level measured in the specific patient on a specific dose to the usual levels produced by the usual therapeutic dose in the drug registration trials. This variation yields the same answer because this comparison takes into account the level in relationship to the dose given in the specific patient versus what occurred in the population of patients in the registration trials. While TDM addresses only variable 2 in equation 1, it does so with much greater quantitative precision than even the most experienced and knowledgeable clinician can do.
Parenthetically, the usual level produced by the usual therapeutic dose is an approximation of the usually effective concentration. This approach minimizes the difficulties encountered in trying to do concentration-response studies in psychiatry. Readers interested in further discussion of this topic are referred to the author’s columns on the rationale for and implications of dose-response curves27 and to Chapter 5 in the Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors.28
Given that TDM simply refines dose titration by taking interindividual variability in clearance into account (equation 2), it is central to the drug development process. As discussed in an earlier column, the goal of drug development is to reduce uncertainty about the effects of drugs.29 The first step in the human phase of drug development (i.e., phase I) is to reduce uncertainty by defining the dose-response curves for the drug and particularly the maximum tolerated dose (MTD). This is initially done in normal volunteers and then may be repeated in mildly symptomatic volunteers.29,30 Once the MTD is defined, that dose is rarely, if ever, exceeded in the rest of the human phases of drug testing. That dose also defines the upper limit of the maximum recommended daily dose range in the package insert for the drug.
Once the drug is marketed, clinicians may be faced with the temptation to exceed the maximum recommended daily dose. This temptation arises when the clinician is faced with a patient who is either incompletely responsive or fully nonresponsive to the new drug. In such cases, the clinician may question whether the maximum recommended dose in the package insert is in fact adequate for a specific patient. As is clear from this discussion, the dose might not be adequate because the patient may be unusually efficient at clearing the drug and therefore has not achieved a sufficient level for response (i.e., the "too low" hypothesis). Alternatively, the patient may be unusually inefficient at clearing the drug and the lack of response is despite (or because) of achieving unusually high concentrations (i.e., the "too high" hypothesis). The clinician may chose to test the "too low" hypothesis by giving an empirical trial of a dose above that recommended in the package insert. Alternatively, the clinician could use TDM to get data relevant to both the "too low" and the "too high" hypotheses as well as compliance by measuring the level of the drug achieved in the patient on the maximum recommended dose and comparing it to the expected concentration reported in the registration trials for the drug.31 The clinician could then use that information to make further clinical judgments about what to do. In essence, if a clinician chooses to exceed the maximum recommended dose, he or she is doing research but in an n = 1 scenario without the usual controls. In this situation, it is difficult to assess with certainty the consequences of such doses, particularly if the adverse consequences of high levels of the drug can mimic worsening of the patient’s illness, as occurred in the case reviewed here.
The question of how "routine" TDM should be in psychiatric practice is beyond the scope of this column and has been addressed elsewhere.32 Instead, the goal is to point out how TDM in specific instances can play a useful role in optimizing medical management by providing objective data about the appropriateness of a dose for a given patient and thus aiding rather than replacing clinical judgment.
In summary, the overall goal of this column, like others in this series, is to illustrate the value of a scientific and disciplined approach to the practice of clinical psychopharmacology to enhance the likelihood of positive treatment outcomes and avoid untoward outcomes.
References
- Preskorn SH, Polypharmacy in a patient with refractory major depression: Part I. The case. Journal of Psychiatric Practice 2002;8:370-6
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Charney DS, Mihic SJ, Harris RA, Hypnotics and sedatives. In: Hardman JG, Limbird LE. Goodman & Gilman?s the pharmacological basis of therapeutics, 10th edition. New York: McGraw-Hill; 2001:399-428
- Physicians’ Desk Reference. Package insert for propranol. Montvale, NJ: Thomson PDR; 2003
- Preskorn SH, Jerkovich GS, Central nervous system toxicity of tricyclic antidepressants: phenomenology, course, risk factors and the role of therapeutic drug monitoring. J Clin Psychopharmacol. 1990;10:88-95
- Bertilsson L, Geographical/interracial differences in polymorphic drug oxidation. Current state of knowledge of cytochromes P450 (CYP) 2D6 and 2C19 Clinical Pharmacokinetics 1995;29:192-209
- Glassman AH, Johnson LL, Giardina EG, et al, The use of imipramine in depressed patients with congestive heart failure. JAMA 1983;250:1997-2001
- Keller MB, Klerman GL, Lavori PW, et al, Treatment received by depressed patients. JAMA 1982;248:1848-55
- Appleton WS, Fourth psychoactive usage guidelines. J Clin Psychiatry 1982;42:12-27
- McLellan RA, Oscarson M, Seidegard J, et al, Frequent occurrence of CYP2D6 gene duplication in Saudi Arabians. Pharmacogenetics 1997;7:187-91
- Teh LK, Ismail R, Yusoff R, et al, Heterogeneity of the CYP2D6 gene among Malays in Malaysia. J Clin Pharm Ther 2001;26:205-11
- Agundez JA, Gallardo L, Ledesma MC, et al, Functionally active duplications of the CYP2D6 gene are more prevalent among larynx and lung cancer patients. Oncology 2001;61:59-63
- Ingelman-Sundberg M, Implications of polymorphic cytochrome P450-dependent drug metabolism for drug development. Drug Metab Dispos 2001;29(4 Pt 2):570-3
- Preskorn SH, Modern drug development and the Human Genome Project. (collection of 4 columns published in the Journal of Psychiatric Practice). Accessed June 25, 2001
- Burrows GD, Norman TR, Tricyclic antidepressants: Plasma levels and clinical response. In: Burrows GD, Norman TR, eds. Psychotropic drugs; Plasma concentration and clinical response. New York: Marcel Dekker; 1981: 69-204
- Asberg M, Individualization of treatment with tricyclic compounds. Med Clin North Am 1974;58:1083-91
- Alexanderson B, Sjoqvist F, Individual differences in the pharmacokinetics of monomethylated tricyclic antidepressants: Role of genetic and environmental factors and clinical importance. Ann NY Acad Sci 1971;179:739-51
- Gonzalez FJ, Skoda RC, Kimura S, et al, Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442-446
- Gonzalez FJ, Vilbois F, Hardwick JP, et al, Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22 Genomics 1988;2:174-9
- Kimura S, Umeno M, Skoda RC, et al, The human debrisoquine 4-hydroxylase (CYP2D) locus: sequence and identification of the polymorphic CYP2D6 gene, a related gene, and a pseudogene. Am J Hum Genet 1989;45:889-904
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Preskorn SH, The slippery slide. J Pract Psychiatry Behav Health 1999;5:50-5
- Preskorn SH, Do you believe in magic? J Pract Psychiatry Behav Health 1997;3:99-103
- Preskorn SH, What happened to Tommy? J Pract Psychiatry Behav Health 1998;4:363-7
- Preskorn SH, Clinical pharmacology case conference: A suicide attempt? Journal of Psychiatric Practice 2002;8:306-10
- Preskorn SH, Antipsychotic drug development in the pre-human genome era: A full circle. Journal of Psychiatric Practice 2001;7:209-213
- Preskorn SH, The rationale for and clinical implications of dose-response curvese project. (collection of columns published in J Pract Psychiatry Behav Health).
- Preskorn SH, Therapeutic drug monitoring. In: Preskorn SH, Clinical pharmacology of selective serotonin reuptake inhibitors. Caddo, OK: Professional Communications; 1996: 99-105.
- Preskorn SH, The stages of drug development and the human genome project: Drug discovery. Journal of Psychiatric Practice 2000;6:341-4
- Preskorn SH, Bridging the gap.
- Preskorn SH, A tale of two patients. J Pract Psychiatry Behav Health 1999;5:160-164
- Burke MJ, Preskorn SH, Therapeutic drug monitoring of antidepressants: Cost implications and relevance to clinical practice. Clinical Pharmacokinetics 1999;37:147-65
|



