|
|
| Reproducibility of the In Vivo Effect of the Selective Serotonin Reuptake Inhibitors on the In Vivo Function of Cytochrome P450 2D6: An Update (Part I) |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice Vol 9, No. 2, March 2003, 150-158
|
The goal of this column is to bring science to the practice of psychiatry through an improved understanding of the mechanisms underlying response to psychiatric medications. The following two equations are central to this understanding:
Effect = affinity for site of action
x concentration at site of action
x biological variance among patients
(genetics, age disease, environment)
(Equation 1)
Drug concentration = dosing rate / clearance
(Equation 2)
There is a reciprocal relationship between all three variables in Equation 1. This fact is particularly obvious with the first two variables: Drug concentration determines which of the drug’s potential sites of action will be engaged and to what degree. Conversely, the affinity of the drug for a site of action determines the concentration of the drug that must be achieved to affect that site of action to a physiologically relevant degree.
The third variable in Equation 1 refers to the biological differences among patients that can shift their position on the usual dose-response curve, thus making them more or less responsive to the drug (i.e., "sensitive" or "resistant").1 Such differences may be mediated through site(s) of action and/or through the mechanisms of pharmacokinetics.
As illustrated in Equation 1, biological differences among patients may be genetically determined (i.e., mutations in the gene[s] coding for the site[s] of action or the gene[s] coding for the mechanisms mediating the absorption, distribution, metabolism or elimination characteristics of the drug). They may also be due to disease- related changes in either the site of action or the pharmacokinetics of the drug. An example of a disease-related change in site of action is the loss of dopamine neurons in the brains of patients with Parkinson’s disease, which makes these patients more sensitive to the extrapyramidal side effects of dopamine antagonists (e.g., haloperidol). An example of a disease-related change that affects the pharmacokinetics of a drug is the slowing of drug clearance caused by a reduction in left ventricular stroke volume, which in turn leads to a reduction in hepatic and renal blood flow and hence drug clearance. There are also age-related changes in physiology that can affect these variables. Finally, there are internal environmental changes that result from the ingestion of substances, which, by virtue of their presence in the body, can alter the response to another drug. These substances may be dietary (e.g., grapefruit juice), herbs (e.g., St. John’s Wort), or drugs (e.g., antidepressants).
A considerable amount of research effort has focused on the fact that some antidepressants under usual dosing conditions are capable of inhibiting one or more human drug metabolizing enzymes and can thus cause substantial elevations in the blood levels of specific coprescribed drugs. That work initially caused some confusion and controversy concerning the reproducibility of these effects and their clinical implications.2 This column will provide an update concerning the effects of the selective serotonin reuptake inhibitors (SSRIs) on one specific human drug metabolizing enzyme, cytochrome P450 (CYP) 2D6. For a more detailed discussion of these issues and those reviewed in the sections that follow, readers are referred to two publications by the author3,4 and the chapter on antidepressants by Shad and Preskorn in Metabolic Drug Interactions.5
CYP 2D6 is one of the group of CYP human drug metabolizing enzymes responsible for the oxidative metabolism essential to the clearance of most drugs (i.e., the denominator in equation 2). This fact is essential in understanding the clinical implications of either inhibiting or inducing such enzymes.
Inhibition leads to a reduction in enzyme activity and thus a decline in the clearance of drugs dependent on that enzyme for their biotransformation to more polar substances, a critical step in the elimination of most drugs. Conversely, induction leads to an increase in enzymatic production and hence activity and thus an increase in clearance of drugs dependent on that enzyme for their biotransformation and elimination.
If the dosing rate remains the same and clearance is reduced, the drug level will increase; the reverse is true for induction (Equation 2). The clinical consequence of the change in drug concentration is dependent on its magnitude and the nature of the drug affected (i.e., "victim").
If the affected or victim drug has a narrow therapeutic index (i.e., a small difference between what is a therapeutic versus a toxic dose), then the result can be catastrophic.6-9 If the change in clearance is modest or the therapeutic index of the drug is wider, then the outcome may simply be a greater or smaller drug effect, making the patient more or less sensitive to the drug.
The latter scenario is more subtle, more common in practice, and harder to detect than is a catastrophic outcome. It can nevertheless be clinically meaningful. That is because the changes that drugs produce in patients’ physiology can create new symptoms rather than producing an improvement in existing symptoms. These new symptoms can be mistaken for worsening of the disease being treated or the emergence of a new disease.10-12 For this reason, such drug-drug interactions can ironically lead to the use of more medicines or other treatments, as the prescriber attempts to treat the new or worsened condition without realizing that it is being caused by a drug-drug interaction.
To put this discussion in perspective, it is important to realize that phase II and III clinical trials are essentially population pharmacokinetic studies in which the goal is to determine the usual dosing rate needed relative to the usual clearance (i.e., clearance in the majority of patients enrolled in a clinical trial) to achieve a concentration that engages the right target to the right degree to obtain the desired benefit-to-risk ratio (i.e., excellent efficacy, no toxicity, and minimal to no tolerability problems).
Cytochrome P450 Enzymes
The cytochrome P450 enzymes are expressed predominantly in the liver but also in other organs such as the bowel wall. These enzymes form a barrier between the contents of the "sewer" that flows through the body (i.e., the inside of human gastrointestinal tract, which parenthetically is outside of but surrounded by the human body). The original function of these enzymes was to protect against the intact absorption of xenobiotic (i.e., foreign biological) toxins that plants produce to protect themselves from being eaten by animals. Since many early drugs, including herbal remedies, were either plant products or derivatives, these drugs were also metabolized by the same enzymes--which is why the CYP enzymes determine the clearance of so many drugs as well as herbal products.
They are referred to as cytochrome enzymes because they contain a heme molecule, which is essential to the electron transfer inherent in the oxidative reactions they mediate. The P stands for pigment since these enzymes were first deduced to exist based on researchers’ observation of pigments in formaldehyde fixed and stained liver tissue using the light microscope. The fact that the production of some of these enzymes could be increased (i.e., induced) was deduced from the observation that the amount of this pigment could be increased by the pretreatment of the animal with drugs such as phenobarbital prior to the preparation of the liver slice for microscopic study. The designation, 450, comes from the wavelength of light absorbed by these enzymes.
| Figure 1. The nomenclature system for cytochrome P450 enzymes based on the degree of homology in the enzymes’ amino acid sequences | 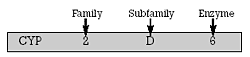 |
CYP 2D6 was the first CYP enzyme that was isolated and characterized. This accomplishment occurred in 1988 through molecular biology techniques, which permitted the identification of the gene which coded for this CYP enzyme. It had been known since the late 1960s that approximately 5%-10% of Caucasians (and smaller percentages of other ethnic groups) were genetically deficient in this enzyme and thus experienced increased frequency and severity of side effects when treated with "usual" doses of drugs dependent on this enzyme for their clearance. Individuals deficient in functional CYP 2D6 have been termed "poor metabolizers" (PMs) to distinguish them from the majority of individuals with functional enzyme, who are termed "extensive metabolizers" (EMs). PMs will achieve levels of CYP 2D6 model substrate/drugs four or more times higher than EMs. By comparing the DNA from individuals with and without this enzymatic activity, the gene coding for the functional enzyme could be identified. Since that time, the genes coding for most, if not all, the human drug metabolizing enzymes have been identified. The identification and study of these genes and their products have led to a rational classification system based on the degree of homology in the enzymes’ amino acid sequences (Figure 1). The CYP enzymes are first grouped into families, which are designated by the first Arabic numeral. All enzymes in a family have at least 40% amino acid sequence homology. They are further grouped into subfamilies designated by an alphabet letter. All enzymes in the same subfamily have at least 55% amino acid sequence homology. The second Arabic numeral designates the gene within the subfamily that codes for a specific enzyme.3
Impact on the Drug Development Process
The isolation of these genes also led to a number of other developments, including the ability to test patients for genetically determined deficiencies in these enzymes (variable 3 in equation 1). Although these tests have generally not yet been incorporated into clinical practice, that is likely to happen in the future. Another development has been the ability to transfect these genes into single cell organisms such as E. coli and thus "humanize" them so that they become capable of expressing one human drug metabolizing pathway.13,14 Such organisms are now used in the drug development process to determine how humans are likely to metabolize a new candidate drug before it is ever given to humans. In fact, that information will partly determine whether a new drug will enter clinical testing. For example, the drug is less likely to be developed if it is principally dependent on an enzyme that is polymorphic in humans, such CYP 2D6 and CYP 2C19.
These transfected organisms can also be used to determine whether a new candidate or an already marketed drug is capable of inhibiting a specific human drug metabolizing enzyme under usual dosing conditions. In this instance, a drug that is normally metabolized by the enzyme expressed by the transfected organism is incubated with the organism and the new candidate drug is added to determine whether it is capable of inhibiting the reaction. The inhibition rate constant of the drug for this reaction can be determined and compared to its kinetic rate constant for its desired effect on its putative mechanism of efficacy (variable 1 in equation 1). If the two rate constants are close or if the drug inhibits the enzyme even more potently than it binds to its desired target, then it is likely it will inhibit that enzyme under dosing conditions needed for clinical benefit. This technology is now part of the "high throughput" screening done to determine what drugs will be taken further into development.
Ironically, as discussed in an earlier column,15 several popular antidepressants, including three SSRIs (fluoxetine, fluvoxamine, and paroxetine), would likely not be developed today because their ability to inhibit one or more than one human drug metabolizing enzyme would be picked up through such screening prior to human testing.
Does It Happen in Humans?
At this stage, the reader may well have thought of the following important questions: In vitro studies are fine but does such inhibition actually occur in humans under clinically relevant dosing conditions? If so, how reproducible is the effect and what is its magnitude?
In vivo studies paralleling those described above with transfected single cell organisms can also be done in humans. In these studies, the participants are analogous to the transfected cell in the test tube in the sense that they contain the enzyme of interest. Like the humanized cell in the test tube, they must be given a specific dose of a drug, which is a model substrate for that enzyme. A model substrate is a drug whose clearance is usually dependent exclusively on that enzyme. The concentration of the drug achieved on the specific dose given is thus a measure of the clearance of the drug:
Clearance = dosing rate / drug concentration
(Equation 2 rearranged)
It is also a measure of the functional activity of the enzyme mediating the biotransformation, which is the rate-limiting step for its clearance. In essence, these studies are in vivo human enzymology.
There are a number of drugs that can and have served as model substrates for CYP 2D6. These include dextromethorphan (a cough suppressant), desipramine (an antidepressant), metoprolol (a beta blocker), and perphenazine (an antipsychotic). In these studies (Table 1), the model substrate/drug is given to the participants in a dose that is sufficiently low so that even marked inhibition of its clearance will not cause severe toxicity. Classically, the individual is allowed to come to steady-state conditions on the model substrate/drug alone and its concentration is measured. That measurement of concentration, which is usually the area under the plasma concentration-time curve (AUC) following a dose, establishes the baseline value (AUC 1). Then, the potential enzyme inhibitor is added to the treatment regimen, along with continued administration of the model substrate/ drug at the same dose as before. The two drugs are typically administered long enough for the potential inhibitor to come to steady-state, since the magnitude of the inhibition is a function of the concentration of the inhibitor. The AUC of the model substrate/drug is then measured again (AUC 2) and compared with the baseline value (AUC 1). The degree of enzyme inhibition is thus quantified using the following equation:
Enzyme inhibition (%) = (AUC2 - AUC1) / AUC1
(Equation 3)
In these studies, participants are first screened to determine that they have a functional copy of the CYP 2D6 enzyme. That can be done either using phenotyping and/or genetic tests. In the phenotyping tests, the individual is a given a dose of a CYP 2D6 model substrate/ drug and then either a blood or urine sample is collected after a specified period of time and analyzed for the parent drug and its metabolite(s) produced via CYP 2D6 mediated biotransformation. PMs will have a markedly higher ratio of the parent drug to the metabolite compared to EMs because they do not have a functional copy of the enzyme to mediate the biotransformation. If this phenotyping approach is used, it is generally confirmed using a genetic test. Since PMs do not have a functional copy of the enzyme, they are excluded from studies examining whether a drug is capable of inhibiting CYP 2D6 mediated metabolism, since the drug cannot inhibit something that is not present.
In addition to determining how much the coprescribed drug on average increases the level of the CYP 2D6 model substrate/drug (i.e., [AUC2 - AUC1] / AUC1), the EMs can also be tested using the phenotyping approach described above to determine how many (i.e., what percentage) were converted into phenocopies of PMs status (i.e., no functional activity of the enzyme) as a result of treatment with the potential CYP 2D6 inhibitor. That result is reported as EM conversion to PM status (see final column in Table 1). This approach will be discussed further in the next column (Part II).
| Table 1. Formal in vivo studies of the effects of different SSRIs on CYP 2D6 model substrates | | SSR1 | Author | N | SSRI Treatment:
dose (mg/day) x
duration (days) | Substrate | Substrate
dosing | (AUC2-AUC1) /
AUC1
(% increase) | EMs
to
PMs | | Citalopram | Gram et al. 1993 | 8 | 40 x 10 | Desipramine | Single dose | 47% | | Citalopram package
insert (PI)17 | NA | 40x22 | Metoprolol | NA | 100% | | | Escitalopram | Escitalopram PI18 | NA | 20 x 21 | Metoprolol | NA | 82% | | | Escitalopram PI18 | NA | 20 x 21 | Desipramine | NA | 100% | | | Fluoxetine | Alfaro et al. 199919 | 8 | 60 x 8* | Dextromethorphan | Single dose | | 63% | | Amchin et al. 200120 | 12 | 20 x 28* | Dextromethorphan | Single dose | | 25% | | Bergstrom et al 199221 | 6 | 60 x 8* | Desipramine | Single dose | 640% | | | 6 | 60 x 8* | Imipramine | Single dose | Imipramine 235%
Desipramine 430% | | | Preskorn et al. 199422 | 9 | 20 x 21 | Desipramine | 21 days | 380% | | | Otton et al. 199323 | 19 | 37 ± 17 x 21 | Dextromethorphan | Single dose | | 95% | | Maes et al. 199724 | 11 | 20 x 28 | mCPP | 7 days | 820% † | | | Alfaro et al. 200025 | 12 | 60 x 8 | Dextromethorphan | single dose | | 42% | | Fluvoxamine | Alfaro et al. 199919 | 6 | 100 x 8 | Dextromethorphan | Single dose | | 0% | | Spina et al. 199326 | 6 | 100 x 10 | Desipramine | Single dose | 14% | 0% | | Paroxetine | Alderman et al. 199727 | 17 | 20 x 9 | Desipramine | 9 days | 421% | | | Brosen et al. 199328 | 9 | 20 x 8 | Desipramine | Single dose | 364% | 78% | | Albers et al. 199629 | 10 | 30 x 4 | Imipramine | Single dose | Imipramine 74%
Desipramine 327% | | | Alfaro et al. 199919 | 8 | 20 x 8 | Dextromethorphan | Single dose | | 50% | | Ozdemir et al. 199730 | 8 | 20 x 10 | Perphenazine | Single dose | 595% | | | Alfaro et al. 200025 | 12 | 20 x 8 | Dextromethorphan | Single dose | | 83% | | Hemeryck et al. 200031 | 8 | 20 x 6 | Metoprolol
(racemic) | Single dose | S-metoprolol 700%
R-metoprolol 410% | | | Sertraline | Alderman et al. 199727 | 17 | 50 x 9 | Desipramine | 8 days | 37% | | | Jann et al. 199532 | 4 | 50 x 7 | Desipramine | 7 days | 0% | | | Preskorn et al. 199422 | 9 | 50 x 21 | Desipramine | 21 days | 23% | | | Solai et al. 199733 | 13 | 50 x >5 | Nortriptyline | Chronic dosing | 14% | | | Ozdemir et al. 199834 | 21 | 94 ± 26 x 24 ±17 | Dextromethorphan | Single dose | 0% | 0% | | Sproule et al. 199735 | 6 | 108 ± 49 x 21 | Dextromethorphan | Single dose | 6% | 0% | | Alfaro et al. 199919 | 7 | 100 x 8 | Dextromethorphan | Single dose | | 0% | | Kurtz et al. 199736 | 6 | 150 x 8 | Imipramine | Single dose | 68% | | | 6 | 150 x 8 | Desipramine | Single dose | 54% | | | Zussman et al. 199537 | 13 | 150 x 29 | Desipramine | Single dose | 70% | | | Alfaro et al. 200025 | 12 | 100 x 8 | Dextromethorphan | Single dose | | 0% | Abbreviations: AUC = area under the plasma concentration–time curve; EM = extensive metabolizer; PM = poor metabolizer; PI = package insert from Physician’s Desk Reference; NA = not available; mCPP = metachlorophenylpiperazine (a metabolite of nefazodone and trazodone)
*60 mg/day for 8 days is a loading dose strategy used to approximate the plasma levels of fluoxetine and norfluoxetine achieved under steady-state conditions on a dose of 20 mg/day.
† 820% is based on all the data. If the two highest increases are excluded, the average was 270%.
Table adapted from Preskorn 1998.2 Copyright Preskorn 2003. |
| Figure 2. Time course for the effect of fluoxetine on CYP 2D6 function using desipramine as the probe drug. Adapted with permission from Preskorn et al. 1994.22 | 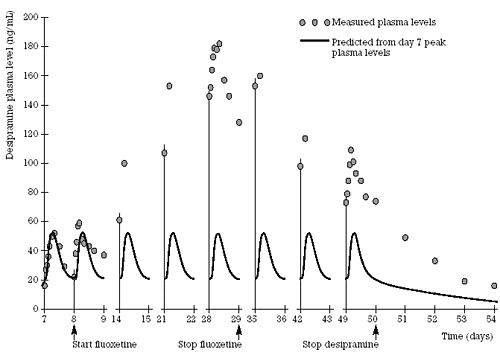 |
Figure 2 shows the data that were generated from a study reporting on the average increase in the AUC of the CYP 2D6 model substrate/drug, desipramine, as a result of coadministration of the SSRI and CYP 2D6 inhibitor, fluoxetine. It also shows the time course of the inhibition of CYP 2D6 as reflected by the increase in plasma concentrations of desipramine. The figure also illustrates the time course of the reversal of the effect following the discontinuation of the inhibitor fluoxetine. While inhibition is an immediate phenomenon unlike induction (which involves increasing the production of the enzyme), the magnitude of the inhibition is a function of the concentration of the inhibitor. If the inhibitor has a long half-life, as is the case with fluoxetine, it will take a prolonged period of administration to achieve its full degree of inhibition and a comparable period of time for full reversal of the inhibition. In Figure 2, the full inhibitory effect of fluoxetine on CYP 2D6 was not measured because, even after 3 weeks of administration, the levels of fluoxetine and its active metabolite, norfluoxetine were still rising. That is also reflected in the fact that even 3 weeks after the discontinuation of fluoxetine, desipramine levels had not returned to baseline because fluoxetine and norfluoxetine had not fully cleared and thus CYP 2D6 inhibition had not been fully reversed.
It should be noted that the average half-life of norfluoxetine is 1-2 weeks in healthy young individuals but 3 weeks in healthy elderly individuals 65 or older.38 Thus, on average it would take 4 months (i.e., 3 weeks x 5 = 15 weeks) to achieve full inhibition of CYP 2D6 in a healthy elderly patient started on 20 mg/day of fluoxetine. It would also take 4 months to achieve full resolution of CYP 2D6 inhibition after fluoxetine was discontinued in such an individual. The issue is not CYP 2D6 inhibition as an abstraction but the change in the CYP 2D6 mediated clearance of coprescribed drugs and hence their accumulation in the body. Parenthetically, the same time course would apply to the other effects of fluoxetine, such as the serotonin uptake inhibition that is presumed to mediate the antidepressant effects of fluoxetine as well as a number of its adverse effects and its potential to pharmacodynamically interact with other serotonin agonists such an monoamine oxidase inhibitors (e.g., to cause the serotonin syndrome).12
Due to the long half-lives of fluoxetine and norfluoxetine, a number of drug-drug interaction studies have employed a loading dose strategy in which 60 mg/day of fluoxetine was administered for 6 to 8 days to achieve an approximation of the concentrations that would be achieved if 20 mg/day had been administered for 5-10 weeks.
Summary of Studies and Results
Table 1 shows the results of the various studies done with citalopram (n = 2), escitalopram (n = 2), fluoxetine (n = 7), fluvoxamine (n = 2), paroxetine (n = 7), and sertraline (n = 10). The next to last column shows the degree of enzyme inhibition and the last column shows the percentage of EMs who were converted to PMs.
| Figure 3. Mean percent increase in the concentration of CYP 2D6 model substrate/drugs as a function of co-administration of different SSRIs | 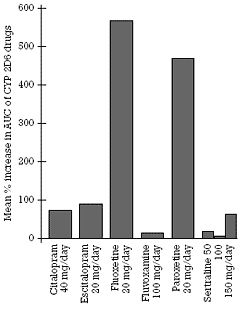 | | Figure 4. Fractional increase in AUC of a model substrate/drug as a function of the percentage decrement in its clearance | 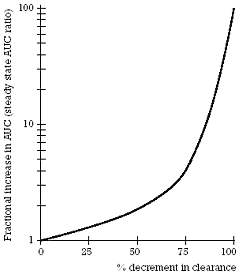 |
As is readily apparent in Table 1, the magnitude of effect seen with each drug is quite similar across studies. In every study, fluoxetine and paroxetine produced substantial inhibition of CYP 2D6 as reflected by the percent increase in the levels of the coprescribed model substrate/drug. In contrast, that was not true for citalopram, escitalopram, fluvoxamine, or sertraline. The average results for each drug are shown in Figure 3.
The reproducibility of the results with the various SSRIs is particularly noteworthy given several considerations. First, various model substrate/drugs were used in a number of these different studies. The fact that comparable results were obtained with different model substrate/drugs indicates that the results can be generalized to other drugs preferentially metabolized by CYP 2D6. Second, the number of subjects in each of these studies was relatively small. The power of this research approach is underscored by the fact that such small numbers can be used to yield highly statistically significant and reproducible results. Third, the variability in the results with fluoxetine and paroxetine is due to the substantial inhibition caused by these drugs and the fact that the curve for the change in clearance as a function of enzyme inhibition is hyperbolic above 70% inhibition, as illustrated in Figure 4. For that reason, modest differences in the degree of inhibition seen between studies can lead to greater than proportional increases in drug concentration (i.e., reductions in clearance).
Only one of the studies with citalopram was published. The other study, like the two studies with escitalopram, have not been published but the results can be found in their respective package inserts. However, a number of details from the studies, such as the number of subjects in each study and their characteristics (e.g., age), are not given in the inserts. The similarity of the results seen with either 40 mg/day of citalopram or 20 mg/day of escitalopram suggests that escitalopram is the active agent mediating the moderate inhibition of CYP 2D6 seen with either the racemate or the single S-enantiomer. This is consistent with the in vitro data showing that S-citalopram is approximately twice as potent as R-citalopram at inhibiting CYP 2D6.39
Sertraline has been the most extensively studied of all the SSRIs in terms of its effects (or absence thereof) on CYP 2D6. It has been studied at three different doses. Fluoxetine is the only other SSRI to have been studied at more than one dose. Whereas the studies done with the other SSRIs have all been done in healthy young volunteers, studies have been done with sertraline in patients as well as volunteers and in the elderly as well as young subjects. These studies all demonstrated modest inhibition of CYP 2D6 at 50 and 100 mg/day. The effect of sertraline at 150 mg/day was comparable to that seen with 40 mg/day of citalopram and 20 mg/day of escitalopram. These results are in contrast to the substantial inhibition seen with 20 mg/day of either fluoxetine or paroxetine.
Summary
As can be readily seen in Figure 3 and Table 1, substantial inhibition of CYP 2D6 is not a "class effect" of the SSRIs, even though this claim was made in the past.2,37 In contrast to fluoxetine and paroxetine, citalopram, escitalopram, fluvoxamine, and sertraline do not produce substantial inhibition of this CYP enzyme under usual antidepressant dosing conditions. While this result is itself important because of its relevance to antidepressant selection and the coprescribing of other medications, the major take-home message of this column is to illustrate how such in vivo studies are done and the reproducibility of the results. In the next column (Part II), the issue of the conversion of EMs to PMs as a result of coadministration of a substantial CYP 2D6 inhibitor will be discussed in more detail.
References
- Preskorn SH, Section 3. The rationale for and clinical implications of dose-response curves. (five columns published in the Journal of Practical Psychiatry and Behavioral Health in 1996)
- Preskorn SH, Debate resolved: there are differential effects of serotonin selective reuptake inhibitors on cytochrome P450 enzymes. J Psychopharmacol. 1998;12:S89-S97
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Shad MU, Preskorn SH, Antidepressants. In: Levy R, Thummel KE, Trager W, Hansten PD, Eichelbaum M, eds. Metabolic Drug Interactions - Drugs as Inhibitors of Metabolic Enzymes Treatment of CNS Diseases. Philadelphia, Pa: Lippincott, Williams & Wilkins. In press
- Preskorn SH, I don’t see ’em. J Prac Psych Behav Hlth. 1997; 3:302-307
- Preskorn SH, Case 1. Sudden death in a 36-year-old man. In Outpatient management of depression. Caddo, OK Professional Communications; 1999:184-6
- Preskorn SH, Clinical pharmacology case conference: A suicide attempt? Journal of Psychiatric Practice 2002;8:306-10
- Preskorn SH, Fatal drug-drug interaction as a differential consideration in apparent suicides. Journal of Psychiatric Practice 2002;8:233-8
- Preskorn SH, Cases 2-4: In Outpatient management of depression. Caddo, OK: Professional Communications; 1999: 186-90
- Preskorn SH, Do you believe in magic? J Pract Psychiatry Behav Health 1997;3:99-103
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Preskorn SH, The stages of drug development and the human genome project: Drug discovery. Journal of Psychiatric Practice 2000;6:341-4
- Preskorn SH, Drug development in psychiatry and genomics: From E. coli to man. Journal of Psychiatric Practice 2001;7:415-9
- Preskorn SH, The human genome project and modern drug development in psychiatry. Journal of Psychiatric Practice 2000;6:272-276
- Gram LF, Hansen MGJ, Sindrup SH, et al, Citalopram: interaction studies with levomepromazine, imipramine, and lithium. Ther Drug Monit. 1993;15:18-24
- Citalopram package insert. Physicians’ Desk Reference. Montvale, NJ: Thomson PDR; 2003: 1344-7
- Escitalopram package insert. Physicians’ Desk Reference. Montvale, NJ: Thomson PDR; 2003: 3532-5
- Alfaro CL, Lam YWF, Simpson J, et al, CYP2D6 Status of extensive metabolizers after multiple-dose fluoxetine, fluvoxamine, paroxetine or sertraline. J Clin Pharmacol 1999;19: 155-63
- Amchin J, Ereshefsky L, Zarycranski W, et al, Effect of venlafaxine versus fluoxetine on metabolism of dextromethorphan, a CYP2D6 probe. J Clin Pharmacol 2001;41:443-51
- Bergstrom RF, Peyton AL, Lemberger L, Quantification and mechanism of the fluoxetine and tricyclic antidepressant interaction. Clin Pharmacol Ther. 1992;51:239-248
- Preskorn SH, Alderman J, Chung M, Harrison W, Messig M, Harris S, Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J Clin Psychopharmacol. 1994;14:90-98
- Otton SV, Wu D, Joffe RT, Cheung SW, Sellers EM, Inhibition by fluoxetine of cytochrome P-450 2D6 activity. Clin Pharm Therap. 1993;53:401-409
- Maes M, Westenberg H, Vandoolaeghe E, et al, Effects of trazodone and fluoxetine in the treatment of major depression: Therapeutic pharmacokinetic and pharmacodynamic interactions through formation of meta-chlorophenylpiperazine. J Clin Psychopharmacol 1997;17:358-64
- Alfaro CL, Lam YWF, Simpson J, et al, CYP2D6 Inhibition by fluoxetine, paroxetine, sertraline, and venlafaxine in a crossover study: Intraindividual variability and plasma concentration correlations. J Clin Pharmacol 2000;40:58-66
- Spina E, Pollicino AM, Avenoso A, Campo GM, Perucca E, Caputi AP, Effect of fluvoxamine on the pharmacokinetics of imipramine and desipramine in healthy subjects. Ther Drug Monit. 1993;15:243-246
- Alderman J, Preskorn S, Greenblatt D, et al, Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 1997;17:284-91
- Brøsen K, Hansen JG, Nielsen KK, Sindrup SH, Gram LF, Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol. 1993;44:349-355
- Albers LJ, Reist C, Helmeste D, Vu R, Jang SW, Paroxetine shifts imipramine metabolism. Psychiatry Research. 1996; 59:189-196
- Ozdemir V, Naranjo CA, Herrmann N, et al, Paroxetine potentiates the central nervous system side effects of perphenazine: Contribution of cytochrome P4502D6 inhibition in vivo. Clin Pharmacol Ther 1997;62:334-47
- Hemeryck A, Lefebvre RA, De Vriendt C, et al, Paroxetine affects metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin Pharmacol Ther 2000;67:283-91
- Jann MW, Carson SW, Grimsley SR, Erikson SM, Kumar A, Carter JG, Lack of effect of sertraline on the pharmacokinetics and pharmacodynamics of imipramine and its metabolites. Clin Pharm Therap. 1995;57:207. Abstract
- Solai LK, Mulsant BH, Pollock BG, et al, Effect of sertraline on plasma nortriptyline levels in depressed elderly. J Clin Psychiatry 1997;58:440-3
- Ozdemir V, Naranjo CA, Hermann N, et al, The extent and determinants of changes in CYP2D6 and CYP1A2 activities with therapeutic doses of sertraline. J Clin Psychopharmacol 1998;18:55-61
- Sproule B, Otton SV, Cheung SW, et al, CYP2D6 inhibition in patients treated with sertraline. J Clin Psychopharmacol 1997; 17:102-6
- Kurtz D, Bergstrom RF, Goldberg MJ, et al, The effect of sertraline on the pharmacokinetics of desipramine and imipramine. Clin Pharmacol Ther 1997;62:145-56
- Zussman BD, Davie CC, Fowles SE, et al, Sertraline, like other SSRIs, is a significant inhibitor of desipramine metabolism in vivo. Br J Clin Pharmacol. 1995;39:550-551
- Harvey AT, Preskorn SH, Fluoxetine pharmacokinetics and effect on CYP2C19 in young and elderly volunteers. J Clin Psychopharmacol 2001;21:161-6
- Von Moltkte LL, Greenblatt DJ, Giancarlo GM, et al, Escitalopram (S-citalopram) and its metabolites in vitro: Cytochromes mediating biotransformation, inhibitory effects, and comparison to R-citalopram. Drug Metab Dispos 2001;19:1102-9
|



