|
|
| To Monitor or Not to Monitor, II: The Glass Is More Than Half Full |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, September 1996, 307-310
|
In law, there are several different levels of proof: probable cause, preponderance, clear and convincing, and beyond a reasonable doubt. In interpreting the results of therapeutic drug monitoring (TDM in most areas of medicine and certainly in psychiatry, we are generally at the level of clear and convincing for toxicity and at the level of preponderance for efficacy. In other words, the glass of evidence is somewhat more than half full when it comes to TDM and efficacy.
In this column, I extend my discussion of the role of TDM in psychiatry to the issue of efficacy (For more discussion of TDM, see my columns in the March and May 1996 issues). I will explain why we are at the level of a preponderance of the evidence and why we are unlikely to go beyond this level until we have made significant advances in our diagnostic sophistication and ability to measure physiologically meaningful markers of clinical response. I will also discuss why correlational approaches to test for a relationship between drug level and efficacy are virtually doomed to fail in psychiatry and why they lead to a false negative conclusion (i.e., a relationship does not exist when in fact it does).
WHAT IS A CORRELATIONAL STUDY?
In a correlational study, the goal is to determine how much of the variability in treatment outcome is a function of variability in the independent variable (plasma drug levels) (Figure 1). In a TDM study, this hypothesis is examined by plotting response as a function of level using each patient as a separate data point. The idea is that patients who fall below a minimum threshold level will have less response than those above.
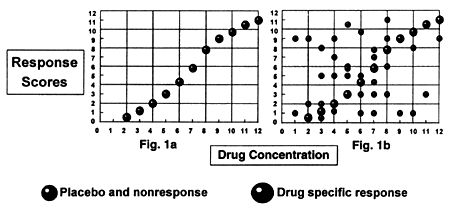 | | Figure 1 - How a poor signal-noise ratio can hinder the ability to establish a concentration-response relationship |
The ideal design for such a study involves the random assignment of patients to treatment with a fixed dose(s) of the medication. Plasma levels will vary as a function of interindividual differences in elimination rates. Occasionally, the study will have more than one fixed dose to which the patient may be assigned. That approach helps to increase the variability of the levels that will be achieved by different patients as a function of treatment.
There are many similarities between these studies and standard clinical efficacy trials. Patients must meet standard diagnostic criteria for whatever condition is being studied, (e.g., major depression, schizophrenia). Improvement in the condition is typically assessed by the same types of rating instruments (e.g., Hamilton Depression Rating Scale). The major difference is that plasma samples are obtained on a predetermined schedule to measure the drug level achieved. Rating scales are completed at the same time to assess clinical response (i.e., change in symptom severity). These two sets of data are then plotted and correlational statistics performed to test for a relationship.
The results of such studies have been modest at best. In many cases, no statistical relationship between response and plasma levels has been found. Such negative results are cited as evidence that no such relationship exists. However, failure to find a relationship using this approach is not surprising, but rather predictable given our current state of knowledge. It does not prove that a relationship between drug levels and response does not exist but simply that we cannot demonstrate it using such an approach.
SIGNAL-TO-NOISE PROBLEMS IN CORRELATIONAL STUDIES
The fact that there have been as many positive studies as there have been is actually amazing, given the inherent signal-to-noise problems caused by the substantial nonspecific treatment response seen in most efficacy studies in psychiatry. (For a detailed discussion of nonspecific treatment response, see my May 1996 column). That there have been positive correlation studies is in part the basis for the subtitle of this column, "the glass is more than half full."
In antidepressant studies, we have the rule of thirds: one third respond to "placebo" treatment, one third respond specifically to drug treatment, and one third do not respond. To illustrate the problem, let us assume that there is a perfect relationship between response and drug level in those patients who respond solely because of the medication (Figure 1a). The problem is that over half the patients who respond on drug in a clinical trial are not responding because of the drug but for other reasons (i.e., nonspecific treatment responders). In other words, specific drug responders = all drug responders - "placebo" responders) In addition, the number of patients who do not respond on drug regardless of the dose and hence plasma drug level (i.e., the final third) is equal to the number who respond specifically because of the drug (i.e., the middle third).
The correlational studies I have been discussing include both nonresponders and "placebo" responders as well as drug-specific responders; however, there is no way to identify the former two groups and suppress their data, which represent "noise" and make it virtually impossible to detect the relationship between response and drug level (figure 1b). The problem is that, although response is not a function of drug treatment in "placebo" treatment, the response data from "placebo" responders will be plotted as a presumed function of their plasma drug level. The same is true for patients who have a form of the illness which is fundamentally not responsive to the mechanism of action of the specific drug and hence would not respond regardless of the plasma drug level achieved.
ASSESSMENT ISSUES IN CORRELATIONAL STUDIES
The other problem in such studies is the Y-axis, which indicates a change in symptom severity as assessed by a rating scale. Such ratings reflect a subjective assessment of a cluster of symptoms, some of which may be core symptoms of the illness while others are not. These scales reflect physiology only indirectly and suffer from being ordinal rather than having a true zero and equal intervals (e.g., the absence of a true zero is the reason that a score somewhere between 7 and 10 on the Hamilton Depression Rating Scale is considered to represent remission). An example of the unequal nature of the intervals is that a drop of four points on such a scale is not necessarily of equal clinical significance depending on where on the scale the drop occurs (e.g., a drop from 32 to 28 versus 18 to 14). We may also interpret the clinical significance differently depending on the types of changes involved (e.g., a four point drop solely due to improvement in sleep versus a four point drop due to improvement in mood and reduced suicidal ideation). In contrast to the Y-axis, the X-axis (i.e., plasma drug level) does have a true zero and equal intervals.
In a correlational study, these nuisances are typically ignored and the response is simply plotted as a function of drug levels. We then endeavor to find how much the variance in one accounts for variance in the other. The scale problem alone would be a challenge, but the situation becomes virtually impossible due to the "signal-to-noise" problem caused by "placebo" responders and drug nonresponders.
SO WHAT TO DO?
Because the goal of the TDM study is to determine whether there is a relationship between drug level and response in patients who are capable of responding specifically to the drug, a simple solution would be to enroll only patients who will respond specifically to the drug. The simplest outcome would be a step function in which there is no response below a critical minimal threshold value and a full response above. As I have discussed in earlier columns, there may also be a critical maximal threshold value above which the risk of toxicity clearly outweighs any likelihood of additional therapeutic benefit. Recall that the definition of a therapeutic range is the drug concentration that is associated with the maximum benefit in the majority of patients without significant risk of adverse effects.
The problem with the simple solution of enrolling only drug-specific responders is that we do not have a reliable means of identifying these patients. Moreover, attempts to reduce the number of "placebo" responders enrolled in such studies have also been generally unsuccessful. As discussed in my last column (July 1996), the Heisenberg principle comes into play here in the sense that the more energy we put into the system the more we change it. Attempts have included setting inclusion criteria that require higher severity and longer duration thresholds for the current depressive episode, but some have wondered whether this leads to a version of "grade inflation." Another failed approach has been to have a longer single-blind placebo run-in phase. However, the longer placebo run-in may actually work against keeping the drug-specific responders in the study, since they may discontinue because of failure to respond. Thus, we must concede that the correlational design will not be an appropriate way to establish the relationship between drug levels of psychiatric medications and their efficacy until we have a truly effective way to identify drug-specific responders. Instead, we must use another research approach.
That approach is to take a page out of the standard clinical trials development program. Such studies provide data to support a determination of whether a drug is sufficiently safe and effective to be approved for marketing. In such studies, the drug is compared to a placebo control and sufficient numbers are enrolled to statistically overcome the "noise" created by not being able to eliminate "placebo" responders and drug nonresponders. The study is a success if it demonstrates that the drug is statistically superior to the "placebo" control condition and the magnitude of the difference in response is clinically meaningful. That result does not require that only patients treated with drug get better but rather that statistically more patients on drug than on "placebo" get better. In fact, some patients treated with "placebo" will get as well as any patients on drugs and some drug treated patients will do as poorly as any patients on "placebo". In a positive study, the difference between the two treatment conditions is such that the drug treated group is skewed towards response while the "placebo" treated group is skewed towards nonresponse. (Figure 2).
| Figure 2 - Estimated minimum effective drug concentration is the average plasma drug concentration achieved in the group treated with the minium, effective dose. | 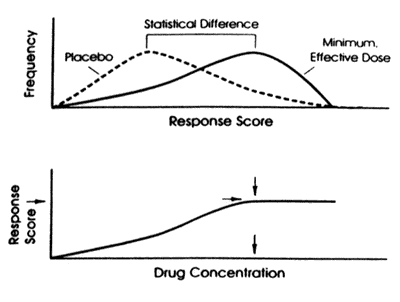 |
This same type of study can also be used to estimate the therapeutic concentration range of the drug if it employs a fixed rather than a titratable dose design. Ideally, such studies will include a dose that is ineffective and several different doses that are effective. For example, selective serotonin reuptake inhibitors (SSRIs) as a class have flat dose-response curves (i.e., there is no increase in the number of responders at doses above the usually effective minimum dose). The ineffective dose and the usually effective minimum dose in such a study define the minimum effective threshold concentration range since each dose determines an expected concentration range in the population being studied (i.e., the age, gender, and ethnic mix of the study population). This concept is illustrated in Figure 2.
SSRIs as a class have a flat dose-antidepressant response curve, meaning response to SSRIs follows a simple step function. Once their mechanism of action is sufficiently engaged to produce an antidepressant response, there is no advantage on average in engaging it more by increasing the amount of the drug. In other words, the relationship between response and plasma drug level plateaus above the critical minimum threshold range. This phenomenon is expected for drugs such as SSRIs which apparently have a single mechanism of action. If a drug has more than one mechanism of action and has different potencies for these different mechanisms of action, further efficacy might be obtained by increasing the concentration above the minimally effective threshold and thus adding another mechanism of action to the therapeutic mix.
Fixed dose studies with SSRIs suggest that efficacy may actually decrease at higher doses (and hence concentrations) because the magnitude of the effect (i.e., the difference between drug and "placebo") decreases at higher fixed doses. This is due to the increased early dropout rate in patients randomly assigned to higher versus lower fixed doses. Using a last observation carried forward analysis, these early dropouts decrease the efficacy seen at higher doses because they stop treatment before they have a chance of responding and thus count as treatment failures. Thus, these higher doses can be used to define an upper concentration range where adverse effects on average exceed therapeutic benefit in most patients. WHY ESTABLISH THE THERAPEUTIC RANGE?
The reader may wonder whether there is any value in knowing the therapeutic range in addition to the therapeutic dose of a drug; however, there are both important clinical and research advantages to defining the therapeutic range.
Clinical Advantages
To understand the clinical value, we must remember that the clinician is primarily interested in what happens to the individual patient rather than to groups of patients. For example, when I present the flat dose-antidepressant response data for the SSRIs, I am almost invariably asked by a clinician what to do for the patient who does not respond to the usually effective minimum dose. This is precisely the situation in which knowing the usually effective threshold plasma drug level can be helpful.
As I mentioned above, a dose defines an expected range of plasma drug levels in the population studied. There is a mean and a standard deviation for that range. One third of patients will be more than one standard deviation either above or below that mean. More than one standard deviation below may be sufficient to lead to non-response and more than one standard deviation above may be sufficient to cause adverse effects that outweigh the therapeutic benefit.
TDM allows the clinician to determine whether the patient is an outlier relative to the expected concentration on the prescribed dose either due to interindividual differences in clearance or noncompliance. If the patient is an outlier, they may not have responded to the usually effective minimum dose because that dose in that patient did not produce the appropriate plasma drug level to maximize the likelihood of an optimum response. TDM provides data indicating whether the problem is too little or too much drug (rather than simply assuming that an inadequate response must mean an inadequate dose). These data can then guide the clinician's decision to try a higher or lower dose.
Issues of Cost
Ironically, one of the most common arguments mounted against TDM in psychiatry is that it is not cost effective; however, the opposite may well be the case. When the cost argument is made, generally only the cost of the TDM is considered, not the costs this test can save. These savings include the cost of failed trials of medications because the dose was not appropriate, the cost of extra physician visits attempting to titrate the dose using clinical assessment of response, the cost of treatments for adverse effects because the dose was too high, and the cost associated with a longer duration of illness as a result of a failed trial due to the use of an inappropriate dose. Using TDM, the physician can reduce these costs by increasing the likelihood that the patient will receive an optimal trial of the medication the first time (i.e., by adjusting the dose if the patient falls outside the range associated with optimal response in most patients in terms of a balance of efficacy and safety/tolerability).
Balanced against all these potential savings is the one-time expenditure for an assay that usually runs less than $50.00. It is usually a one-time expenditure because TDM measures a patient's intrinsic ability to clear a specific drug. For most drugs, this ability is a trait phenomenon that does not change without reason (the only exceptions to this general rule are lithium and carbamazepine due to their more complicated pharmacokinetics). For all other psychiatric medications, TDM is repeated only for cause, most commonly suspected noncompliance or a significant change in the patient's status that would be reasonably expected to alter the ability to clear the drug (e.g., impairment in heart, liver, or renal function due to disease or the addition of a concomitant medication that can affect cytochrome P450 enzyme function [see my November 1995 column for a discussion of these enzymes]).
Obviously, a sizable percentage of patients will respond to the usually effective dose, but there is also a percentage who would benefit from a dose adjustment. The issue of when to use TDM boils down to how large this latter group is and the cost of doing TDM relative to the savings that will be realized. If the group that would benefit from a dose adjustment based on TDM is relatively small, then the savings might not offset the cost of TDM for the entire group. In this situation, it would be more cost-effective to give an adequate trial of the usually effective dose and reserve TDM for the nonresponder group. If the nonresponder group is relatively large, then it might be more cost-effective to use TDM as a one-time
standard of care in the entire population early in treatment. This would ensure that everyone receives a dose adjusted for their clearance rate that will produce optimal drug levels.
Research Advantages
Establishing the therapeutic concentration range for a drug also has research implications. It can help establish the clinically relevant mechanism of action of the drug. While the putative mechanism of action underlying clinical efficacy is typically inferred from preclinical research, knowing the usually effective concentration can help support or disprove this hypothesis. If the putative mechanism is not likely to be meaningfully engaged at the concentration that is usually effective, then that suggests the need to search for other possible mechanisms of action. Conversely, the putative mechanism of action is supported by the finding that the usually effective minimum concentration of the drug is sufficient to meaningfully engage the suspected mechanism of action. For example, the finding that the average plasma drug level of each SSRI produced by its respective, usually effective, minimum dose for antidepressant efficacy is sufficient to cause 70% inhibition of serotonin uptake is strong support for this mechanism being clinically relevant to the antidepressant properties.
In the next column, I will discuss our emerging understanding of pharmacogenetic variability in end organ responsitivity and how such variability may shift the dose response curve in specific patients.
Suggested Readings
- Preskorn SH. Clinical pharmacology of selective serotonin reuptake inhibitors. Caddo, OK: Professional Communications, Inc. 1996.
- Preskorn S, MacD.: The implication of concentration response studies of tricyclic antidepressants for psychiatric research and practice. Psychiatric Developments 1984; 3:201-22
|



