|
|
| Bupropion: What Mechanism of Action? |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, January 2000, 272-276
|
This column is the fourth in a series discussing how to assess the potential relevance of in vitro binding studies to the clinical use of a drug. The series has used the various classes of antidepressants as examples to illustrate basic pharmacologic principles. In the previous columns, I have focused on antidepressants that are serotonin uptake pump inhibitors (SRIs), the selective serotonin reuptake inhibitors (SSRIs), and venlafaxine. In this column, I will discuss bupropion, which has an in vitro and clinical pharmacology substantially different from that of a serotonin uptake pump inhibitor.
There are two caveats to this column. The first caveat is that I have a special fondness for bupropion because of our intertwining history. The major milestones in that history are described below.
First, bupropion launched my research experience in clinical trials. I was a principal investigator on clinical trials to test its antidepressant efficacy in the late 1970s. Of interest, the target doses in those early studies were between 600 and 900 mg/day. Those doses are 1.5-2.0 times the currently recommended maximum dose.
Next, I served (with full disclosure of my research activities) on the neuropsychopharmacology advisory committee of the Food and Drug Administration that reviewed the new drug application for bupropion and recommended it for approval. Those hearings were held in the early 1980s. It may surprise some readers to know that, except for a series of miscues, bupropion could have been marketed before fluoxetine. A discussion of those miscues is beyond the scope of this column, but they delayed the marketing of the drug for over a year after it was deemed approvable. During that delay, the Pope study found a high incidence of seizures (8%) in a relatively small group of patients with bulimia (data on file with Food and Drug Administration). This finding caused Burroughs-Wellcome to literally recall the trucks that were delivering the drug to pharmacy shelves.
I later served as a consultant to the company when it re-applied for approval to market bupropion with a cap on the maximum recommended daily dose. Finally, I worked with Burroughs-Wellcome as a consultant as they endeavored to develop a sustained release formulation of bupropion to minimize the seizure risk and extend the patent life of the drug.
The second caveat to this column is that there are a number of important unknowns about the clinically relevant pharmacodynamics and pharmacokinetics of bupropion. For this reason, the conclusions in this column are often best inferences based on what is known. These inferences could serve as hypotheses for further studies. The large number of unknowns with bupropion is a direct result of its history. First, bupropion is relatively old in terms of drug development in the United States.1 In some respects, it is older than fluoxetine. For that reason, much of the technology and the requirements now in place to study the pharmacodynamics and pharmacokinetics of central nervous system drugs did not exist when bupropion was developed. Generally, companies do not invest in such studies after a drug has been approved unless the FDA or the market requires it. Parenthetically, psychiatry as a field has generally not been proactive in requiring companies to do studies to clarify the pharmacology of drugs once they have been marketed. Second, companies are more likely to invest in studies when a drug is doing well in the market. That simply makes good business sense. Bupropion sales languished for a number of years for reasons that are beyond the scope of this column. However, its sales have substantially improved since it received approval as an aid to smoking cessation. Nevertheless, bupropion's pharmacology database reflects its age and postmarketing status. With these caveats in mind, let us proceed to consider the potential clinical relevance of the in vitro pharmacology of bupropion.
The binding affinities of bupropion
Figure 1 shows the binding affinity of bupropion for a variety of neural sites of actions with potential relevance to clinical pharmacology (i.e., capable of mediating either desired or undesired effects) as well as the binding affinities for the same neural sites of action of three other types of antidepressants: amitriptyline (an antidepressant with multiple mechanisms of action), desipramine (a norepinephrine selective reuptake inhibitor [NSRI]), and sertraline (an SSRI).4,5
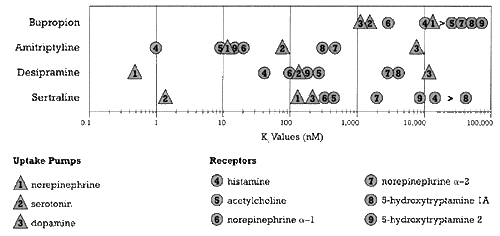 | Figure 1 - Relative Potency for Different Sites of Action of bupropion and three other antidepressants.
- Based on data from Bolden-Watson C, Richelson E, 1993, Cusack B, et al. 1994 |
Sertraline is arguably the best SSRI to contrast with bupropion because it has the highest binding affinity for the dopamine uptake pump of any SSRI and any antidepressant.2,6-8 We can thus compare and contrast sertraline and bupropion to make the point that in vitro binding affinity, while important, is only one of three factors that determine drug effect as expressed in the following equation:
(Equation 1)
Effect = affinity for x drug x biological
site of action level variance
As is readily apparent from Equation 1, binding affinity does not tell the clinician what a drug will do under clinically relevant dosing conditions, but rather how much drug is needed to produce a specific effect (i.e., what drug level is needed to engage a site of action to a physiologically meaningful degree). Binding affinity or potency is a concentration-dependent term.5 Drug level in turn determines the dosing rate that is needed in relationship to the patient's ability to clear the drug. This relationship is expressed in the following equation:
(Equation 2)
Drug level = Dosing rate / Clearance
I stress this theme because it is so basic to understanding the clinical effects of any drug.
What is the neural mechanism of action of bupropion?
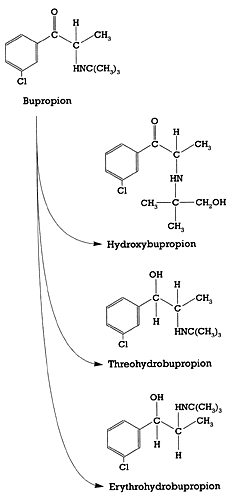 | Figure 2 - Structure of bupropion and its three major metabolites
- Preskorn 199114 |
The pivotal observation about bupropion from Figure 1 is that this drug does not bind avidly to any of the common neural sites shown in the figure. This observation suggests one of two conclusions: 1) the clinical effects of bupropion are not mediated through any of these sites of action, or 2) high enough concentrations of bupropion are reached under usual antidepressant dosing conditions to affect one or more of these sites to a physiologically significant degree. The weight of the evidence suggests that the second explanation is correct, but with a twist: It is not just bupropion that is active but also its metabolites.9-12
Of all the possible sites of action shown in Figure 1, bupropion binds most avidly to the dopamine and norepinephrine uptake pumps . The adverse effect profile of bupropion is consistent with the conclusion that treatment with this drug produces physiologically relevant inhibition of these two pumps under usual antidepressant dosing conditions.13 Specifically, two of bupropion's most common adverse effects are tremors and sweating, which are consistent with effects as a dopamine and norepinephrine agonist, respectively.13
To find out how this effect is possible given the weak affinity of the drug for these targets, we need to look at the pharmacokinetics of bupropion. The parent drug is biotransformed by multiple CYP enzymes into several metabolites: hydroxybupropion, erthyrobupropion, and threohydrobupropion (Figure 2). Although the relative contribution of each of these metabolites to the antidepressant or epileptogenic effects of bupropion has not been established, they are pharmacologically active.9-12 Moreover, because of their slower clearance, these metabolites accumulate to a much greater extent than the parent drug.15,16 The total concentration of bupropion plus these metabolites far exceeds the concentration of any antidepressant except nefazodone (Table 1). Thus, the total concentration of bupropion plus its major metabolites is consistent with the relatively low affinity for the dopamine and norepinephrine uptake pumps.
So why do we postulate that bupropion affects the dopamine uptake pump and sertraline does not?
Based on the data in Figure 1 alone, one might draw the following faulty conclusion: that sertraline, being more potent that bupropion as an inhibitor of dopamine uptake, must inhibit the dopamine uptake pump under its usual antidepressant dosing conditions if bupropion does. I discussed the reasons why we conclude sertraline most likely does not inhibit this pump under its usual antidepressant dosing conditions in an earlier column and will not repeat that discussion here. However, a discussion of the pharmacokinetic differences between bupropion and sertraline should help to illustrate why in vitro pharmacology cannot be interpreted without an appreciation of the pharmacokinetics of the drugs.
In contrast to bupropion, the principal metabolite of sertraline is much less potent in terms of its binding affinity for any of the neural sites shown in Figure 1.6 Moreover, its concentration is on average only 1.5 times higher than that of the parent drug.6 For these reasons, this metabolite is unlikely to contribute in any appreciable way to the pharmacology of sertraline, at least with respect to the sites shown in Figure 1.
Tables 1 and 2 provide the data needed to understand why bupropion affects the dopamine and norepinephrine uptake pumps while sertraline does not. The differences between the plasma levels of sertraline and those of bupropion plus its metabolites occur because of rather than in spite of the substantial difference in the binding affinity of bupropion and sertraline for their respective targets. The levels of sertraline are so low because of its high binding affinity for the serotonin uptake pump. In contrast, the level of hydroxybupropion alone is 100 times higher than the level of sertraline because such high concentrations are needed to block the dopamine and norepinephrine uptake pumps, given the low affinity of bupropion for these targets.
| | | Table 1. Mean (and standard deviation) trough plasma levels (ng/ml) of bupropion and its three major metabolites in patients on 450 mg/day of immediate release forumulation.* | | Bupropion | 33 | (34) | | Hydroxybupropion | 1452 | (666) | | Erythrohydrobupropion | 138 | (86) | | Threohydrobupropion | 671 | (425) |
| | * Part of the data in this table was previously published in Preskorn and Katz 1989.16 |
| | Table 2. Relationship between sertraline dose, mean trough plasma level and magnitude of serotonin uptake inhibition in platelets.* | | Dose | 50 mg | | Mean drug level | 16 ng/ml | | SE uptake inhibition of platelets | 80% |
| | * Table modified from Preskorn 1999.18 |
|
Based on the comparison of binding affinities shown in Figure 1, the levels of sertraline needed to block the dopamine uptake pump are clearly lower than the levels of bupropion needed to block this pump. However, the serotonin uptake pump would be saturated at such concentrations of sertraline. In an earlier column,19 the dose-(and hence concentration-) dependent nature of sertraline's inhibition of the serotonin uptake pump was presented. Based on those results, a dose of 200 mg/day in the average patient produces sertraline concentrations that yield over 95% inhibition of the serotonin uptake pump. Given the difference in sertraline's binding affinity for the serotonin versus the dopamine uptake pump, the dose of sertraline needed to block the dopamine uptake pump would be substantially higher than 200 mg/day. Nevertheless, this statement is tentative since there have been no formal, human studies done to determine what dose of sertraline would be needed to achieve concentrations of sertraline sufficient to block dopamine uptake nor how many people could tolerate such a dose in terms of the serotonin adverse effects likely to result from such a saturation of the serotonin uptake pump.
The bottom line is that the 20-fold difference between sertraline and bupropion in binding affinity for the dopamine uptake pump (Figure 1) is more than offset by the 100-fold difference in the drug concentrations of each achieved under their usual dosing conditions (Tables 1 and 2). Parenthetically, notice that the difference in concentration between these two drugs is several times higher than the difference in their dose (50 to 450 mg/day, respectively). Remember that dose is only one of the two factors that determine drug concentration (Equation 2). That is the reason why dose is an imperfect way of assessing the potency of a drug in terms of an action.
Do these issues relate to the seizure risk on bupropion?
Quite likely. The reason for the equivocation is that so little is known about the pharmacology underlying the epileptogenic properties of bupropion. We do not know whether the risk is due principally to the parent drug or to one of its three major metabolites or to a combination of more than one of the four.
Given this dearth of knowledge, how could this discussion be relevant to the risk of seizures on bupropion? First, an axiom of pharmacology is that some mechanism must underlie the epileptogenic effects of bupropion. Blockade of an ion channel is a good possibility as a potential mechnism mediating this effect since such a mechanism is common with other epileptogenic drugs. Second, the seizure risk with bupropion is clearly concentration dependent given the risk factors summarized in Table 3. If we knew the affinity of bupropion for the responsible site of action, we could plot it in Figure 1 and give a visual representation of the therapeutic index that exists with this drug (i.e., the difference in concentration needed for efficacy versus that needed to cause this untoward effect).
The high concentrations of bupropion and its metabolites needed for antidepressant efficacy (Table 1) set the stage for its narrow therapeutic index. The explanation is as follows: The low affinity of bupropion goes against the modern tenets of drug development--which is to try to produce drugs that have high binding affinity for the site(s) of action mediating their efficacy. Such a development strategy makes it possible to produce a drug with a wide separation between its binding affinity for the site mediating efficacy and its affinity for sites mediating adverse effects. That wide separation in binding affinity means a wide therapeutic index (i.e., the difference between the lowest dose or floor concentration needed for efficacy and the upper dose or ceiling concentration at which concerns about adverse effects outweigh clinical benefit).4 The high concentrations of bupropion and/or its metabolites needed for efficacy establish a high floor concentration and thus set the stage for a narrow therapeutic index.
| Table 3. Factors suggesting that high levels of bupropion and/or its metabolites are linked to the occurence of seizures.* | - Incidence of seizures is a function of dose, either
quantitated as total daily dose or mg/kg/day dose. - Most seizures occur within the first several days of a
dose increase. - Most seizures occur within the first several hours
after taking the dose. - Patients with anorexia or bulimia nervosa, who have
lean body mass and the potential for rapid drug
absorption have an increased risk of seizures.
| | * Preskorn 1991,14Davidson 1989,20 |
What is the clinical relevance of the complex metabolism of bupropion?
In this series of columns, I have been focusing on the relationship between the in vitro pharmacology of a drug and its pharmacokinetics. Bupropion is arguably the best CNS example of the importance of this relationship because of the multiple biotransformation pathways involved in its clearance (Figure 2) coupled with the widely varying activities of these pathways in different patients (Table 1). For these two reasons, four different chemicals accumulate to varying but substantial degrees during bupropion treatment. As mentioned earlier, these metabolites are pharmacologically active but their relative contribution to the beneficial and adverse effects associated with bupropion treatment is not known.
Based on several observations, different CYP enzymes are likely to be involved to a clinically significant degree in the biotransformation of bupropion. In vitro drug metabolism studies have shown that several different CYP enzymes (i.e., CYP2B6, 3A4, 2A6, 2E1, and 1A2) can mediate the biotransformation of bupropion.21 However, the fact that an enzyme can mediate a drug's biotransformation in vitro does not axiomatically mean that it will do so in vivo to a meaningful degree under clinically relevant dosing conditions. Instead, the kinetic rate constant for the reaction (like the kinetic dissociation rate constant from an in vitro receptor binding study) must be considered relative to the drug concentration achieved under such dosing conditions. If the drug's affinity for the enzyme is low relative to the concentration achieved, then that enzyme is unlikely to be involved to any meaningful degree in the drug's biotransformation. Unfortunately, the published in vitro drug metabolism studies with bupropion do not provide sufficient detail to estimate the relative contribution of these different CYP enzymes to its biotransformation under clinically relevant dosing conditions.21 Nevertheless, it is likely that more than one enzyme is involved in the biotransformation of bupropion under clinically relevant dosing conditions since the levels of the different metabolites do not correlate with each other. If one enzyme mediated more than one of the pathways, then there should be a correlation between the levels of the products formed by the same enzyme across different patients, since the activity of the enzyme would be the same within a given individual, even though its activity varied across individuals.
The importance of this discussion is obvious when we consider the narrow therapeutic index of bupropion in terms of its concentration-dependent risk of causing seizures. Differences in enzyme activity across individuals can result in a differential accumulation of the parent drug and its metabolites, which in turn could result in a differential response in terms of either beneficial or adverse effects. These differences might exist because of genetic polymorphisms, gender, age, disease, or environmental factors that can induce or inhibit specific CYP enzymes (e.g., carbamazepine or fluoxetine, respectively).
Conclusion
The pharmacology of bupropion provides an excellent illustration of the "take home" messages behind this entire series of columns.
1. First and foremost, physicians cannot rely solely on the results of in vitro studies with a drug to determine what that drug will do in clinical practice. This is true whether the in vitro study is assessing the dissociation rate constant of a drug's binding affinity for a receptor or the kinetic rate constant of an enzyme for a drug. Both of these numbers tell us how much drug (i.e., drug concentration) is needed for that mechanism to come into play (Equation 1). If that fact is not taken into account, erroneous conclusions can result. For example, Figure 1 taken alone could lead one to mistakenly conclude that sertraline is more likely than bupropion to block dopamine uptake under each drug's respective clinical dosing conditions for antidepressant response.
2. This column provides a foundation for understanding the cautions that should be taken in dosing bupropion. These include the need (a) to carefully titrate the dose to achieve the lowest effective dose, (b) to not exceed 450 mg/day, (c) to not exceed 150 mg in any single dose, and (d) to not give doses closer than every 6 hours.22 Whenever a drug's labeling includes a warning to not exceed a specific dose, the prescriber should suspect that the drug has a narrow therapeutic index and wide interindividual variability in drug metabolism. This is true for bupropion. The goal of setting an upper threshold is, in essence, to "cap" the percentage of the population who will achieve a drug concentration that carries the risk of drug toxicity. For further discussion of this issue, the reader is referred to several earlier columns.23-25
The regular reader of this column will also understand why drugs with such dosing guidelines are prime candidates for therapeutic drug monitoring (TDM). The intent behind TDM is to permit rational dosage adjustment to compensate for interindividual differences in drug clearance (Equation 2). Unfortunately, we in clinical psychopharmacology often do not have as much information as we would like to confidently establish TDM-driven dosage guidelines. Bupropion is a classic example. While bupropion's seizure risk is dose- and hence concentration-dependent, we do not know the answers to two important questions. What levels should be measured: bupropion, hydroxybupropion, threohydrobupropion, erythrohydrobupropion, or some combination of these four? And what is the upper threshold beyond which the risk of seizures outweighs the likelihood of further clinical benefit? There are a number of reasons why we do not have this information. However, that discussion is beyond the scope of this column. For now, as a first approximation of upper threshold concentrations, it would seem prudent to not exceed the average levels of bupropion and each of its metabolites that are obtained on the maximum recommended daily dose (i.e., 450 mg/day).
3. Prescribers should reconsider the dose of bupropion when starting or stopping a CYP enzyme inducer or inhibitor, since such an action could change the clearance of bupropion and/or one or more of its metabolites. Such a change could in turn affect either the efficacy and/or safety of bupropion. Again, for all the reasons discussed in this column, there is an unfortunate dearth of information about which enzyme inducers or inhibitors pose a risk. Hence, the prescriber should keep this issue in mind whenever using bupropion with a drug that produces a substantial change in the activity of any drug-metabolizing enzyme (e.g., fluoxetine, carbamazepine).
In my next column, I will discuss three different antidepressants, each of which has multiple mechanisms of action: imipramine, mirtazapine, and nefazodone.
References
- Soroko FE, Mehta NB, Maxwell RA, Ferris RM, Schroeder DH, Bupropion hydrochloride ((+/-) alpha-t-butylamino-3-chloropropriophenone HCI): A novel antidepressant agent. J Pharm Pharmacol 1977;29:767-70
- Bolden-Watson C, Richelson E, Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029
- Cusack B, Nelson A, Richelson E, Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559-565
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Frazer A, Antidepressants. J Clin Psychiatry. 1997;58(suppl 6):9-25
- Hyttel J, Comparative pharmacology of selective serotonin reuptake inhibitors (SSRIs). Nord J Psychiatry. 1993;47 (suppl 30):5-12
- Ascher JA, Cole JO, Colin J-N, et al, Bupropion: A review of its mechanism of antidepressant activity. J Clin Psychiatry 1995;56:395-401
- Cooper TB, Perumal AS, Suchow RF, Glassman A, Bupropion: Possible role of major metabolites in mode of action (Abstract). Clin Pharmacol Ther 1984;37:187
- Martin P, Massol J, Colin JN, Lacomblez L, Puech AJ, Antidepressant profile of bupropion and three metabolites in mice. Pharmacopsychiatry 1990;23:187-94
- Schroeder DH, Metabolism and kinetics of bupropion. J Clin Psychiatry 1983;44(Suppl 5):79-81
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Preskorn SH, Should bupropion dosage be adjusted based upon therapeutic drug monitoring? Psychopharmacol Bull. 1991;27:637-643
- Golden, RN, DeVane CL, Laizure SC, Rudorfer MD, Sherer MA, Potter WZ, Bupropion in depression: The role of metabolites in clinical outcome. Arch Gen Psychiatry 1985;45:145-49
- Preskorn SH, Katz SE, Bupropion plasma levels: Intraindividual and interindividual variability. Ann Clin Psychiatry 1989;1:59-61
- Preskorn SH, De-spinning in vitro data. J Pract Psychiatry Behav Health 1999;5:283-7
- Preskorn SH, Two in one: The venlafaxine story. J Pract Psychiatry Behav Health 1999;5:346-50
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Davidson J, Seizures and bupropion: a review. J Clin Psychiatry. 1989;50:256-261
- Wurm RM, Dunn JA, Silver I, Patel DK, In vitro metabolism of bupropion by human liver microsomes and cDNA expressed human cytochrome P450s. Proceedings of the International Society for the Study of Xenobiotics 1996;10:371
- Physicians’ Desk Reference. Montvale, NJ: Medical Economics; 1999:1252-5
- Preskorn SH, If lack of concentration didn’t cause the fall, what did? J Pract Psychiatry Behav Health 1996;2:364-7
- Preskorn SH, To monitor or not to monitor? J Pract Psychiatry Behav Health 1996;2:172-5
- Preskorn SH, Why did Terry fall off the dose-response curve? J Pract Psychiatry Behav Health 1998;4:363-7
|



