|
|
| Imipramine, Mirtazapine, and Nefazodone: Multiple Targets |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Practical Psychiatry and Behavioral Health, March 2000, 97-102
|
General Bucky Turgeson in the Stanley Kubrick movie, Dr. Strangelove, was mighty proud of his aerial warriors and their B-52s. This series of planes, first rolled out in the 1950s, are still being used. The longevity and value of these planes are the result of their ability to deliver so many different types of payloads to their targets.
For similar reasons, I suspect Bucky would have loved imipramine, mirtazapine, and nefazodone. These drugs are multiple action antidepressants that are capable of affecting multiple targets at the concentrations achieved over their clinically relevant dosing range (Figure 1). That fact distinguishes them from the norepinephrine selective reuptake inhibitors (NSRIs) (e.g., desipramine) and from the selective serotonin reuptake inhibitors (SSRIs) (e.g., sertraline) (Figure 2). NSRIs and SSRIs have a binding affinity for the site believed to mediate their antidepressant efficacy that is at least an order of magnitude greater than their binding affinity for other neural mechanisms of action.5 In this column, I will focus on how the differences between the in vitro pharmacology of the multiple mechanism of action antidepressants (e.g., imipramine, mirtazapine, nefazodone) and that of the single mechanism of action antidepressants (i.e., SSRIs and NSRIs) account for the differences in their clinical pharmacology.
This column is the fifth in a series reviewing the clinical relevance of in vitro binding affinity. Although I have focused on antidepressants in this series, the basic pharmacological principles I have covered are applicable to any class of medications. The two primary principles underlying this series are:
- In vitro binding affinity, while important, is only one factor in the equation that determines drug effect:
(Equation 1)
Effect = affinity for x drug x biological
site of action level variance
Binding affinity does not tell you what a drug will do but rather how much drug is needed to produce a specific effect--in other words, what drug level is needed to engage a site of action to a physiologically meaningful degree. The reason is that binding affinity or potency is a concentration-dependent term.5
- The drug level needed to engage the desired target to a physiologically meaningful degree dictates the dosing rate that is needed relative to the patient's ability to clear the drug. The relationship between drug level, drug dose, and drug clearance is expressed in the following equation:
(Equation 2)
Concentration = Dosing rate / Clearance
In this column, I discuss how the data in Figure 1 relate to the clinical pharmacology of imipramine, mirtazapine, and nefazodone. In doing so, I present a pharmacological explanation for why both imipramine and mirtazapine have curvilinear dose-response curves with regard to some of their actions and why nefazodone has an ascending dose-antidepressant response curve like venlafaxine rather than the flat dose-antidepressant response curve seen with the SSRIs.
Imipramine
In the first column in this series, I made the point that a 10-fold separation in binding affinity between two targets is generally required to be able to achieve a drug concentration that will affect the highest affinity target to a physiologically meaningful degree without affecting the other target .5 From this standpoint, imipramine is the antithesis of a selective drug. Within a concentration range that involves less than a 10-fold separation, imipramine binds to six targets: the uptake pumps for norepinephrine and serotonin and the histamine-1, muscarinic acetylcholine, serotonin 2A (5-hydroxy tryptamine-2A, 5-HT2A), and alpha-1 adrenergic receptors (Figure 1). Blockade of these receptors will cause sedation, dry mouth, and orthostatic hypotension, respectively, whereas inhibition of the uptake pumps is believed to mediate the antidepressant efficacy of these medications.6
 | Figure 1 - Relative potency for different sites of action of non-SSRI antidepressants
- Based on data from Bolden-Watson C, Richelson E, 1993, Cusack B, et al. 1994 and
de Boer et al. 1988, 1995 |
Just as important, although not shown in Figure 1, imipramine also blocks the sodium fast channel at a concentration only slightly more than 10 times that needed to block the norepinephrine uptake pump.6 That fact explains why the tertiary amine tricyclic antidepressants (TCAs) can cause cardiac conduction disturbances and why they have a narrow therapeutic index. The cardiotoxicity of these drugs results from their ability to inhibit fast sodium channels and thus prolong intracardiac conduction.8 This prolongation in turn sets the stage for escape ventricular beats, which can result in a fatal ventricular arrhythmia. The 10-fold separation between imipramine's affinity for the norepinephrine uptake pump and for the fast sodium channels means that an imipramine concentration can be achieved that produces an antidepressant response without causing delayed intracardiac conduction. However, the fact that the separation is only slightly more than 10-fold means that taking an overdose consisting of the entire amount contained in a typical 2-week prescription can result in a fatal arrhythmia.
As I discussed in an earlier column,7 these pharmacological properties are the reason tertiary amine TCAs such as imipramine served as the blueprint for the development of subsequent antidepressants. Studying the pharmacology of drugs like imipramine (Figure 1) and amitriptyline (Figure 2) helped us formulate hypotheses about what sites of action mediated their desired (e.g., antidepressant efficacy) and their undesired (e.g., dry mouth, cardiotoxicity) effects. These hypotheses in turn led to the synthesis of new drugs that mimic only part of the pharmacology of these older antidepressants.
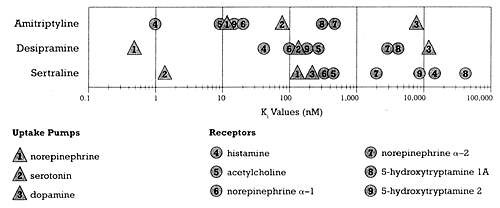 | Figure 2 - Relative potency for different sites of action of three different types of antidepressants.
- Based on data from Bolden-Watson C, Richelson E, 1993, Cusack B, et al. 1994 |
Curvilinear dose-response curves seem paradoxical to many people. They ask how a drug can produce an effect at one dose but not at a higher dose. The answer is that, at lower concentrations (i.e., low dose), the drug preferentially engages a site of action capable of mediating the first effect, but at higher concentrations (i.e., higher doses), the drug engages a second site of action that blocks or antagonizes the first effect. Imipramine has just such a curvilinear dose-response curve with regard to blood pressure.9 At low concentrations, imipramine increases blood pressure by increasing the ionotrophic effect of norepinephrine on the myocardium and its vasoconstrictive effect on peripheral arterioles via its blockade of the norepinephrine uptake pump. Imipramine shares this action with other norepinephrine reuptake inhibitors, including its active metabolites, desipramine, and the serotonin-norepinephrine reuptake inhibitor, venlafaxine.10 However, in contrast to desipramine and venlafaxine, imipramine at higher doses antagonizes its initial pressor effect on the cardiovascular system and can even cause orthostatic hypotension by blocking the alpha-1 adrenergic receptor resulting in decreased peripheral resistance. Since desipramine and venlafaxine do not block the alpha-1 adrenergic receptor over their clinically relevant dosing range, the likelihood and magnitude of their pressor effect simply increase as the dose of these drugs is increased.
Mirtazapine
As with all drugs, knowledge of the in vitro pharmacology of mirtazapine can help us understand its clinical pharmacology. Mirtazapine's most potent site of action is the histamine-1 receptor (Figure 1). At higher concentrations, the drug sequentially blocks the 5-HT2A receptor and then the alpha-2 adrenergic receptor (Figure 1). Although not illustrated in Figure 1, mirtazapine binds almost as avidly to the 5-HT2C and 5-HT3 receptors as it does to the 5-HT2A receptor.3,4
Doses of mirtazapine needed to treat clinical depression result in concentrations sufficient to block the 5HT2A, 5-HT2C, 5-HT3, and the alpha-2 adrenergic receptors. Higher concentrations (and hence doses) are obviously needed to block the alpha-2 adrenergic receptor site than are needed to block the serotonin receptors. These facts are consistent with the hypothesis that one or more of these mechanisms mediate the antidepressant efficacy of mirtazapine. This constellation of mechanisms of action is not shared by any other antidepressant, which raises the possibility that mirtazapine might work in patients who have not benefited from other types of antidepressants. There is some evidence supporting this possibility. Two separate studies have indicated that mirtazapine can work in patients who have not benefited optimally from treatment with amtriptyline11 or fluoxetine.12
Based on its binding profile, some psychiatrists have used mirtazapine both to augment the antidepressant effects of serotonin uptake inhibitors (i.e., SSRIs and low dose venlafaxine) and to treat their adverse effects. To put this discussion in perspective, a few comments about the effects of SSRIs and venlafaxine may be helpful. These antidepressants are believed to work by increasing serotonin availability to the various serotonin receptors in the brain by slowing the neuronal reuptake of serotonin after its release. Thus, these drugs work as indirect serotonin receptor agonists. While SSRIs are "selective" in terms of directly affecting only the serotonin uptake pump, they are not "selective" in terms of their agonism of serotonin receptors. They increase serotonin availability to all serotonin receptors. Agonism of the 5-HT1A receptor is currently believed to be the mechanism mediating the antidepressant efficacy of these drugs. Conversely, agonism of the 5-HT2A, 5HT2C, and 5-HT3 receptors may principally mediate the adverse effects of these drugs: sleep disturbance, anxiety and weight gain, and nausea/loose stools/vomiting, respectively.7
Mirtazapine at antidepressant concentrations blocks the 5-HT2A, 5-HT2C, and 5-HT3 receptors. Thus, the combination of this drug plus an SSRI or venlafaxine could simultaneously treat the adverse effects of these drugs while increasing the availability of serotonin to the 5-HT1A receptor, the putative mechanism mediating their antidepressant efficacy. Mirtazapine also blocks the alpha-2 adrenergic receptor (Figure 1). Thus, mirtazapine could act like yohimbine, another alpha-2 adrenergic antagonist, to treat sexual dysfunction caused by serotonin uptake inhibitors. However, that potentially desirable effect may be offset by its blockade of the histamine-1 receptor (you may simply be too tired to be frisky). Alpha-2 adrenergic antagonism also promotes the release of serotonin. That coupled with serotonin uptake inhibition could increase serotonin availability postsynaptically. That could either be beneficial (e.g., increased antidepressant efficacy) or adverse (e.g., serotonin syndrome) depending on the magnitude of the increase and at what postysynaptic receptors it occurs.
To address this issue, we conducted a study to determine what happens when patients who have been on the SSRI, fluoxetine, are immediately switched to mirtazapine.12 Fluoxetine was used in this study for two reasons. First, it persists for weeks in the body even after abrupt discontinuation. For that reason, patients switched from fluoxetine to other drugs are actually being treated with both drugs for weeks after the switch, even though they and their physicians may not consider this fact when interpreting their response. Second, due to its inhibition of multiple cytochrome P450 enzymes, fluoxetine might interact both pharmacokinetically and pharmacodynamically with mirtazapine.6
As expected, there was an interval of several weeks when these patients had measurable and pharmacologically relevant plasma concentrations of mirtazapine and fluoxetine plus norfluoxetine. None of the patients developed the serotonin syndrome and their adverse effects were qualitatively and quantitatively the same as those seen in patients on mirtazapine, 15 mg/day, alone. As also expected, fluoxetine did not appreciably alter the concentration of mirtazapine consistent with the fact that mirtazapine has multiple pathways for its elimination.13
Mirtazapine, like imipramine, may have a curvilinear dose-response curve. There is some suggestion that sedation is more pronounced on low rather than high dose mirtazapine therapy (15 versus 30 mg/day or more). This suggestion is principally based on the fact that there was a higher incidence of sedation in the American trials, which used lower doses, than in the European trials, which used higher doses of mirtazapine. There may be other reasons for this finding but that discussion is beyond the scope of this paper. Suffice it to say that the multiple actions of mirtazapine suggest the following theoretical explanation for why this otherwise paradoxical observation might be true. The sedative effect of mirtazapine at low doses is consistent with its high affinity for the histamine-1 receptor. Given that mirtazapine binds more avidly to that site of action than to sites capable of mediating relief from a depressive episode (Figure 1), sedation occurs at doses of mirtazapine below those needed for antidepressant efficacy (i.e., less than 15 mg/day). However, higher doses of mirtazapine result in the blockade of the alpha-2 adrenergic receptor, which produces an alerting or arousal effect just like yohimbine and just the opposite of the sedation produced by the alpha-2 adrenergic agonist, clonidine. Thus, mirtazapine most likely causes sedation at low doses (i.e., concentrations) by preferentially blocking the histamine-1 receptor, while at higher doses (i.e., concentrations), mirtazapine blocks the alpha-2 adrenergic receptor, which theoretically could reduce its sedating effects to some degree. A fixed dose trial or another type of prospective study would be needed to rigorously test this concept.
Nefazodone
The first target affected by nefazodone is the 5-HT2A receptor (Figure 1). As the concentration of nefazodone increases, it sequentially engages the alpha-1 adrenergic receptor and then the serotonin uptake pump (Figure 1). There is almost an order of magnitude difference between nefazodone's binding affinity for the 5-HT2A receptor and its affinity for the serotonin uptake pump. Moreover, nefazodone is converted into a triazolodione metabolite, which is over 100 times more potent as a 5-HT2A receptor blocker than an uptake pump inhibitor.14 While this metabolite is ten times less potent than the parent drug as a 5-HT2A blocker, its trough concentrations under steady-state dosing conditions are ten times higher than that of the parent drug, consistent with its longer half-life (24 versus 4 hours).15 Based on these facts, it should be possible to achieve concentrations of the parent drug nefazodone and its triazolo metabolite that cause substantial and sustained blockade of the 5-HT2A receptor without producing substantial and sustained inhibition of the serotonin uptake pump.
As with the SSRIs and venlafaxine,16 human studies using platelets as a surrogate measure for central serotonin uptake inhibition have been done with nefazodone to examine its dose-dependent effects on this mechanism. Nefazodone at a dose of 300 mg/day for 14 days produces 20%-40% inhibition of serotonin uptake inhibition in comparison to the 70%-80% inhibition produced by the SSRIs and venlafaxine at their respective, lowest usually effective doses.10,17-19
The degree of serotonin uptake inhibition produced by nefazodone, 300 mg/day, relative to the SSRIs and venlafaxine, is consistent with its in vitro binding affinity and its drug concentration. While nefazodone is capable of inhibiting the serotonin uptake pump, it is relatively weak in terms of its binding affinity. For example, nefazodone's binding affinity is three orders of magnitude weaker than paroxetine or sertraline.20 Given that difference, some may find it surprising that nefazodone produces any inhibition of the serotonin uptake pump. It does so for the same reason that bupropion inhibits the dopamine uptake pump at its usually effective dose.21 The reason is that the concentrations of nefazodone achieved within its clinically relevant dosing range are three orders of magnitude higher than the concentrations of the SSRIs (Table 1).10
In fact, the low binding affinity of nefazodone for the serotonin uptake pump may partly explain why such relatively high concentrations of nefazodone are needed to achieve clinical efficacy as an antidepressant.
| Table 1. Peak plasma levels (ng/ml) of nefazodone and its metabolites (mean +/- standard deviation) at different doses. | | Dose | Nefazodone | Hydronefazodone | Triazolodione | Methylchlorpiperazine | | 100 b.i.d.22 | 880 (+/- 417) | 265 (+/- 112) | 1,080 (+/- 303) | 20 (+/- 5) | | 200 b.i.d.23 | 1,723 (+/- 532) | 433 (+/- 95) | N.A. | 33 (+/- 12) | | 300 b.i.d.24 | 2,980 (+/- 805) | 803 (+/- 214) | N.A. | 79 (+/- 74) |
| | * The above values are for men. Young and older men and young women achieve comparable plasma levels on the same dose while elderly women develop 50% higher plasma levels. |
Parenthetically, the same point was made in the preceding column about concentrations of bupropion and its metabolites relative to their binding affinity for the dopamine and norepinephrine uptake pumps.21 Nevertheless, the degree of serotonin uptake inhibition produced by nefazodone, 300 mg/day, is probably not sufficient in and of itself to produce an antidepressant response. The basis for this inference comes from fixed dose studies with citalopram, paroxetine, and venlafaxine, in which it was found that those antidepressants, at doses that should produce less than 70% inhibition of serotonin uptake inhibition, were not superior to placebo in the treatment of patients with major depression.10
Thus, the platelet uptake inhibition studies with nefazodone provide a heuristic explanation for several of the ways in which the clinical pharmacology of nefazodone differs from that of the SSRIs and venlafaxine.
First, at 300 mg/day, nefazodone does not appear to produce as robust an antidepressant response on average as do the lowest usually effective doses of SSRIs and venlafaxine.15 This statement is based on the magnitude of the reduction of depressive severity as measured by the Hamilton Depression Rating Scale (HDRS)25 score in the antidepressant group versus its parallel placebo control group in double-blind, efficacy trials.
Second, nefazodone, like venlafaxine and in contrast to the SSRIs, has an ascending dose-antidepressant response curve.15 The difference is that, at its lowest recommended dose, venlafaxine produces a reduction in HDRS scores that is comparable to that seen with the SSRIs, whereas nefazodone, as already mentioned, does not. When the dose of nefazodone reaches the upper end of its recommended dose range, the reduction is comparable and perhaps even superior to the reduction produced by the SSRIs, but not as great as occurs with the highest dose of venlafaxine.15
Third, low-dose nefazodone treatment causes a lower incidence of sexual dysfunction than occurs on sertraline and most likely the other SSRIs and venlafaxine.26 As is often said, your strength is often also your weakness. Thus, the lower degree of serotonin uptake inhibition produced by low-dose nefazodone treatment relative to the SSRIs and venlafaxine could account for both its decreased antidepressant efficacy and its lower incidence of sexual dysfunction relative to these other antidepressants.
Several inferences can be drawn from these observations. First, the blockade of 5-HT2A receptors (i.e., the predominant action of nefazodone at 300 mg/day) is not as robust an antidepressant mechanism of action as is serotonin uptake inhibition. That inference is further supported by the fact that antidepressant clinical trials programs with more than one relatively pure 5-HT2A blocker have not shown superior efficacy over placebo. Second, the fact that nefazodone at higher doses is comparable and perhaps even superior to the SSRIs in terms of HDRS score reduction suggests that 5-HT2A blockade combined with serotonin uptake inhibition may be superior to either mechanism alone. That may either reflect a synergistic action, or it may be that 5-HT2A blockade reduces adverse effects due to serotonin uptake inhibition that either mimic depressive symptoms (e.g., insomnia) or lead to treatment discontinuation before efficacy is achieved. Conceivably, a drug with a smaller gap between its binding affinity for the 5-HT2A receptor versus the serotonin uptake pump might be more efficacious or better tolerated than either nefazodone or the SSRIs. Such inferences can lead to further discovery work to develop and test such a compound as an antidepressant. Third, these observations indicate why dose titration is more necessary with nefazodone than with most of the SSRIs to obtain an adequate antidepressant response. Unfortunately, the nonlinear pharmacokinetics of nefazodone make such dose titration more difficult than would be expected with a drug with linear pharmacokinetics. Further discussion of this topic is beyond the scope of this column.
Conclusion
Imipramine, mirtazapine, and nefazodone are at the opposite end of the spectrum from the SSRIs (e.g., sertraline) or the NSRIs (e.g., desipramine or reboxetine) in terms of number of mechanisms of action that are affected at concentrations achieved in the usual patient taking these drugs at their respective antidepressant doses. To the best of our knowledge, the behavioral pharmacology of the SSRIs and the NSRIs is constrained to the effects mediated by their single mechanism of action, serotonin uptake inhibition and norepinephrine uptake inhibition, respectively. In contrast, the pharmacology of imipramine, mirtazapine, and nefazodone is richer or more varied or more complicated (i.e., pick the spin you prefer) than that of the SSRIs as a result of their effects on multiple mechanisms of action. That pharmacology includes both desired and undesired effects, which are often mirror images of the same phenomenon. It is for this reason that drug therapy is rarely, if ever, foolproof and why proper education is needed to prescribe and monitor medications.
The goal of this series of columns has been to help clinicians better understand the differences in the in vitro pharmacology of the various antidepressants as they relate to their clinical pharmacology in patients. This knowledge may help clinicians optimize their selection of an antidepressant for a given patient by matching the pharmacology to the symptom profile of the patient. The clinician can also use this knowledge to help guide the choice of an alternative antidepressant if a patient either does not benefit from or does not tolerate treatment with the first antidepressant chosen.
In my next column, I will present tables that summarize the adverse effect profile of the antidepressants that have been reviewed in this series of columns. These tables will further demonstrate how our ability to predict the pharmacology of a drug depends on understanding the relationship between its pharmacodynamics and its pharmacokinetics.
References
- Bolden-Watson C, Richelson E, Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029
- Cusack B, Nelson A, Richelson E, Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559-565
- de Boer TH, Maura G, Raiteri M, de Vos CJ, Wieringa J, Pinder RM, Neurochemical and autonomic pharmacological profiles of the 6-aza-analogue of mianserin, Org 3770 and its enantiomers. Neuropharmacology. 1988;27:399-408
- de Boer T, Ruigt G, Berendsen H, The alpha2-selective adrenoceptor antagonist Org 3770 (mirtazapine remeron) enhances noradrenergic and serotonergic transmission. Hum Psychopharmacol. 1995;10:107S-118S
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Preskorn SH, Irwin HA, Toxicity of tricyclic antidepressants-kinetics, mechanism, intervention: a review. J Clin Psychiatry. 1982;43:151-156
- Preskorn SH, Factors affecting the biphasic concentration: Effect relationships of tricyclic antidepressants. In: Usdin E, Dahl SG, Gram LF Lingjaerde O, eds. Clinical pharmacology in psychiatry. [CITY?] Macmillan; 1981:297-306
- Preskorn SH, Two in one: The venlafaxine story. J Pract Psychiatry Behav Health 1999;5:346-50
- Catterson M, Preskorn SH, Bremner JD, Dessain EC, Double-blind crossover study of mirtazapine, amitriptyline and placebo in patients with major depression. Poster presentation at the 36th annual meeting of the American Psychiatric Association. New York, May 7, 1996
- Preskorn SH, Omo K, Magnus RD, Shad MU, Assessment of potential drug-drug interactions following immediate crossover from fluoxetine to mirtazapine. Poster session, American Psychiatric Association Meeting, San Diego, 1998
- Delbressine L, Vos R, The clinical relevance of preclinical data: Mirtazapine, a model compound. J Clin Psychopharmacol 1997;17:29S-33S
- Taylor DP, Carter RB, Eison AS, et al, Pharmacology and neurochemistry of nefazodone, a novel antidepressant drug. J Clin Psychiatry. 1995;56(suppl 6):3-11
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Eison AS, Eison MS, Torrente JR, Wright RN, Yocca FD, Nefazodone: Preclinical pharmacology of a new antidepressant. Psychopharmacol Bull 1990;26:311-15
- Owens M, Ieni J, The serotonergic antidepressant nefazodone inhibits the serotonin transporter: In vivo and ex vivo studies. Life Science 1995;57:373-80
- Narayan M, Anderson G, Cellar J, Mallison RT, Price LH, Nelson JC, Serotonin transporter-blocking properties of nefazodone assessed by measurement of platelet serotonin. J Clin Psychopharmacol. 1998;18:67-71
- Preskorn SH, De-spinning in vitro data. J Pract Psychiatry Behav Health 1999;5:283-7
- Preskorn SH, Bupropion: What mechanism of action? Journal of Psychiatric Practice 2000;6:39-44
- Barbhaiya RH, Buch AB, Greene DS, Single and multiple dose pharmacokinetics of nefazodone in subjects classified as extensive and poor metabolizers of dextromethorphan. Br J Clin Pharmacol 1996;42:573-81
- Barbhaiya RH, Shukla UA, Kroboth- PD, Greene DS, Coadministration of nefazodone and benzodiazepines:.Il. A pharmacokinetic interaction study with triazolam. J Clin Psychopharmacol 1995;15:320-6
- Barbhaiya RH, Buch AB, Greene DS, A study of the effect of age and gender on the pharmacokinetics of nefazodone after single and multiple doses. J Clin Psychopharmacol 1996;16:19-25
- Hamilton M, Development of a rating scale far primary depressive illness. Brit J Soc Clin Psychol 1967;6:278-96
- Feiger A, Kiev A, Shrivastava RK, Wisselink PG, Wilcox CS, Nefazodone versus sertraline in outpatients with major depression: Focus on efficacy, tolerability, and effects on sexual function and satisfaction. J Clin Psychiatry 1996;57:53-62
|



