|
|
| Antipsychotic Drug Development in the Pre-Human-Genome Era: A Full Circle |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice, May 2001, 209-213
|
This column begins a mini-series that will review the history of antipsychotic drug development in the era before the human genome project (HGP). This mini-series serves several purposes:
- There have been a number of requests to present and discuss the binding affinities of older and newer antipsychotics. In this regard, this series will complement the earlier series on the clinical relevance of the binding affinities of antidepressants.1
- Ziprasidone (Geodon), the fourth of the newer antipsychotics, was marketed this year. The previous three medications in this group were olanzapine (Zyprexa), quetiapine (Seroquel), and risperidone (Risperdal). Ziprasidone may be the last in this pharmacologic group for at least a couple of years. Thus, this series of columns may have some "shelf life" in terms of remaining clinically relevant and complete. Other antipsychotics are currently moving through development, but these agents are pharmacologically distinct from olanzapine, quetiapine, risperidone, and ziprasidone. While predicting which new drug in a therapeutic area is likely to be approved next is like predicting the stock market, aripiprazole is a reasonable bet. Unlike olanzapine, quetiapine, risperidone, and ziprasidone, aripiprazole is a partial agonist at the dopamine D2 receptor.2-3 Consistent with its basic pharmacology, its clinical pharmacology is distinct from the other newer antipsychotics.
- This review of the history of antipsychotics and the previous columns on the antidepressants will provide a useful frame of reference for subsequent columns about changes in the process of psychiatric drug development as a result of the decoding of the human genome.
Coming Full Cycle
Ironically, the development of antipsychotics has come full circle (Figure 1 and Table 1). The first wave of effective antipsychotics was serendipitously discovered in the 1950s and had multiple mechanisms of action.4-5 The second wave (e.g., haloperidol) was rationally developed to have a specific and selective mechanism of action -- dopamine D2 receptor blockade.5 The third wave completed the circle in that the newest antipsychotics also have multiple mechanisms of action; specifically, they combine blockade of certain subtypes of serotonin (5hydroxytryptamine, 5-HT) receptors and blockade of the D2 receptor. In contrast to the first wave, the third wave was rationally designed to have multiple mechanisms of action. This mini-series of columns will present the historical reasons behind this full circle phenomenon and discuss how it reflects the development process in the pre-HGP era.
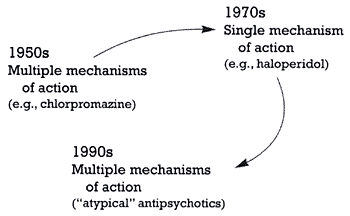 | | Figure 1 - The evolution of antipsychotics |
While the antipsychotics have come full circle, the most popular antidepressants in use today -- the selective serotonin reuptake inhibitors (SSRIs) -- are at the halfway point in this cycle. They represent drugs that were designed to have a single (i.e., selective) mechanism of action.6 From this perspective, fluoxetine (or any of the other SSRIs) is to amitriptyline as haloperidol is to olanzapine (or any of the other newer antipsychotics) in terms of having a single rather than multiple mechanisms of action. As explained in the previous series on antidepressants, one could argue that some of the newer antidepressants -- specifically bupropion, mirtazapine, nefazodone, and venlafaxine-have completed the circle in that they have multiple mechanisms of action.7-9
| Table 1. The three waves of antipsychotics | | | First wave | Second wave | Third wave | | Number of mechanisms of action | Multiple | Single | Multiple | Members of this class
| chlorpromazine
mesoridazine
thioridazine | fluphenazine
haloperidol
molindone | olanzapine
resperidone
quetiapine
ziprasidone | |
One could also say that the various augmentation strategies (e.g., adding pindolol to an SSRI) and combination strategies (e.g., mirtazapine plus venlafaxine) represent an attempt by clinicians to bring antidepressants full circle, since these strategies involve combining drugs with different mechanisms of action to increase efficacy and/or treat adverse effects.10 In contrast to drugs with multiple actions, these augmentation and combination strategies have not undergone the rigorous and extensive clinical testing needed to evaluate their safety or efficacy. The National Institute of Mental Health has recently commissioned the Sequential Treatment Algorithm for Refractory Depression (STAR*D) study to provide prospective (although open) data about the relative efficacy, safety, and tolerability of many of these strategies (http://www.edc.gsph.pitt.edu/stard/index.html-ssi).
The First Wave
Returning to antipsychotics, chlorpromazine (Thorazine) was the first successful antipsychotic. Like many of the first drugs in any therapeutic area (nonpsychiatric as well as psychiatric), chlorpromazine was discovered by chance -- the result of a search by the French anesthesiologist and surgeon, Henri Leborit, for a better pre-anesthetic agent to calm patients going to surgery.11 The researchers called chlorpromazine "Largiticil" because of its multiple actions. Because it lowered blood pressure (a consequence of its ability to block alpha-1 adrenergic receptors), chlorpromazine was abandoned as a pre-anesthetic agent. However, Dr. Leborit hypothesized that a drug that calmed normal people going to surgery might also have anxiolytic effects in psychiatric patients. Thus, chlorpromazine entered clinical testing in patients with a wide range of psychiatric disorders. Through this testing, chlorpromazine was found to have anxiolytic effects in anxious patients but -- more importantly -- it was also found to have rather remarkable beneficial effects in patients suffering from psychotic illnesses, including schizophrenia and bipolar disorder.12 With that discovery, the modern era of antipsychotics was launched.
Parenthetically, chlorpromazine also gave birth to the modern era of antidepressants, since imipramine (Tofranil) was synthesized as a structural analog of chlorpromazine.6,13 The goal was to produce an antipsychotic superior to chlorpromazine. Although imipramine failed as an antipsychotic, it was serendipitously found to have antidepressant effects and thus launched the modern era of antidepressant therapy. The rest of that story was the subject of the earlier series of columns mentioned above.
However, the story of the early antipsychotics and antidepressants illustrates the first pre-HGP approach to psychiatric drug development. In this approach, a drug is discovered by chance and then synthetic chemists tinker with the structure in hopes of coming up with a new chemical entity (NCE)14 that is more effective and/or safer and/or better tolerated. These synthetic chemists then use both their successes and their failures to continually refine their understanding of the relationship between the structure of molecules and their pharmacologic effects (i.e., structure-activity relationships [SARs]).14
This approach has several limitations. First, it is inefficient, being primarily based on trial and error. Second, it is prone to produce drugs with the same mechanism of action, even though it may increase the agent's potency and selectivity for that mechanism. These limitations are in stark contrast to drug development in the HGP era, when a novel regulatory protein can be identified and a drug can then be developed to stereospecifically interact with it.14 That approach is much more target-driven and can lead to the development of truly novel agents. Such a target-driven approach has come to the forefront in psychiatric drug development with the completion of the HGP. The field has not yet seen the fruits of this approach, although it is now being used in drug discovery research. In addition to developing truly novel compounds, this approach may also shave a substantial amount of time off the 10 plus years currently required to develop a new psychiatric drug. Even if this savings in time proves to be true, it will likely be several years before drugs developed based on the HGP make it to the market.
Returning to the history of antipsychotic drug development, synthetic chemists working for a variety of pharmaceutical companies developed a number of other variations on the structure of chlorpromazine. These included thioridazine (Mellaril), mesoridazine (Serentil), and clozapine (Clozaril). It may be surprising to some readers that clozapine is not a new drug. It was synthesized in 1959 and marketed in several European countries in the 1960s.15 However, clozapine is important because it led to the concept of "atypicality," which is now the buzz word when talking about antipsychotics.16,17 Nevertheless, further discussion of "atypicality" will have to wait for a later column in this series. Suffice to say at this time that chlorpromazine begot clozapine which in turn begot olanzapine (the result of synthetic chemists tinkering with the chlorpromazine molecule). The other newer antipsychotics also owe their existence to chlorpromazine.
| Table 2. Chlorpromazine: Polypharmacy in a single pill | | Action | Selective drug with that effect | Histamine-1 blockade
Muscarinic acetylcholine blockade
Serotonin-2A receptor blockade
Dopamine-2 receptor blockade
Alpha-1 adrenergic receptor blockade | chlorpheniramine
benztropine
ritanserin*
haloperidol
prazosin | | | *An investigational antipsychotic whose development was abandoned due to lack of efficacy. |
Chlorpromazine had the same problem as the tertiary amine tricyclic antidepressants-too many unnecessary actions, which in turn produced too many unwanted side effects. Like amitriptyline, chlorpromazine is, in essence, polypharmacy in a single pill (Table 2). The same is true for thioridazine and mesoridazine. These three drugs and related early antipsychotics were grouped together under the label "low-potency" antipsychotics. This term referred to the fact that considerably higher doses of these antipsychotics were needed in comparison to later antipsychotics, such as haloperidol and fluphenazine (Prolixin), to achieve antipsychotic effects.18 In contrast, antipsychotics like fluphenazine and haloperidol were termed "high-potency" antipsychotics based on the dose needed to treat psychosis. They also became known as neuroleptics because of their propensity to cause extrapyramidal side effects (EPS). The high-potency antipsychotics were derivatives of the low-potency antipsychotics. The goal in this second wave of antipsychotic development was to discover drugs that had fewer types of side effects and higher antipsychotic potency. This second wave of antipsychotics (e.g., fluphenazine, haloperidol) will be the subject of another column.
Low Potency Versus "Atypicality"
As mentioned earlier, the first wave of low-potency antipsychotics bears more similarity to the third wave of "atypical" antipsychotics than to the second wave of highpotency antipsychotics. In fact, most of the low-potency antipsychotics fit at least some of the criteria of "atypicality" that have been used to describe the newer antipsychotics. Specifically, many of these low-potency antipsychotics have:
- Greater potency for blocking 5-HT2A than D2 receptors
- Little risk of causing acute EPS
- Low risk of causing tardive dyskinesia and dystonia
- Low risk of causing elevated serum prolactin levels at therapeutic doses
Other properties that have been claimed for "atypicals" include:
- Improvement of deficit symptoms independent of positive symptoms in patients with schizophrenia
- Activity against mood symptoms, both mania and depression
- Improvement of cognitive deficits in schizophrenia
- Activity in patients with neuroleptic-refractory schizophrenia
Many of these latter characteristics were not issues when the low-potency antipsychotics were developed and hence were not studied. However, lack of study does not necessarily mean these drugs do not have these effects. In fact, there are some data that at least some of the low-potency antipsychotics have some of these properties, particularly in terms of activity against mood symptoms. For example, thioridazine is the only antipsychotic formally labeled by the Food and Drug Administration (FDA) for the treatment of clinical depression and anxiety disorders.19 However, that label is an anomaly reflecting the era in which thioridazine was developed rather than the existence of what would now be considered adequate clinical trial data to support such labeling. In other words, the requirements for such a labeled indication have increased since thioridazine was approved.
However, there are double-blind, prospective data showing that chlorpromazine treats acute mania.20 In fact, chlorpromazine was the comparator in some of the lithium trials conducted in the United States in the early 1970s that led to the FDA approval of lithium for mania. In those studies, chlorpromazine was as effective as lithium in controlling acute mania and had a faster onset of action, but was less well tolerated.
Downside to Low-Potency Antipsychotics
While the antipsychotics of the first wave were low potency in terms of antipsychotic effects, they were high potency with regard to antihistaminic and anticholinergic effects.21 Those actions predict adverse effects on cognition -- specifically, impairment of attention, concentration, and memory. Those were significant problems with these drugs and led to the search for the second wave of antipsychotics, the high-potency agents.
Recall that the major goal in drug development is to reduce uncertainty about a drug's effects.14 Much of that reduction is accomplished during the drug discovery and preclinical testing phases. It is further reduced by human testing, first in phase I studies and then in phase II and III studies. In these phases, specific measurements are made to assess the efficacy and safety of the drug. In psychiatric drug development studies, efficacy is generally assessed using researcher-administered and sometimes patient self-report rating scales as well as by global clinical assessments. The latter are generally done using a scale such as the Clinical Global Inventory, which rates the severity of the condition being studied in the patient on a scale from 1 = normal to 7 = most severely ill the clinician has ever seen. Safety measurements included in virtually all clinical trials across all therapeutic areas include routine laboratory tests (e.g., liver function tests, complete blood count), an electrocardiogram, and a physical examination. Other tests may be included for specific reasons based on the pharmacology of the investigational drug or an active comparator drug, Tolerability and unexpected safety problems are also assessed by asking a general question such as: "Have you had any problems since the last time you were seen in the study?" The response to this broad general question is then recorded and includes nuisance adverse events of everyday life, such as headaches, as well as serious, unexpected adverse events, such as heart attacks and automobile accidents. Those events may or may not be related to the drug, but they occurred during treatment and thus could be due to the drug. The principal way causality is tested is by statistically comparing the incidence in the group treated with the investigational drug with the incidence in the group treated with placebo.22 If the incidence rate is higher to a statistically significant degree in the group receiving the investigational drug, then it is concluded that the drug can cause this adverse effect. Parenthetically, the incidence rate is sometimes lower in the group receiving the investigational drug, which may mean that the drug is capable of preventing or treating a common spontaneous adverse event of everyday life. For example, in double-blind, placebo-controlled trials, the reported incidence of headaches is lower in depressed patients treated with imipramine than in those treated with placebo.22
While an attempt is made to fully discover the effects of a new drug during its development, the approach is not foolproof. First, patients may underreport some adverse effects for reasons such as embarrassment, and the researchers may not be aware of the need to specifically inquire about such adverse effects. This phenomenon is the most likely reason that the incidence of sexual dysfunction on SSRIs was underreported in the early clinical trials of these agents. Another reason an adverse effect may be missed is that it occurs too infrequently (i.e., less than 1 in 100 but more than 1 in 1,000) or too rarely (i.e., less than 1 in 1,000) to show up as a statistically significantly greater risk on investigational drug versus placebo. Most clinical trial programs involve exposing about 3,000 humans to the drug during its phase I through phase III testing. That fact is the simplest reason that some adverse effects are discovered only after the drug has become available on the market. The number of people exposed to a drug increases by orders of magnitude even within a few weeks of marketing. If the adverse effect is infrequent or rare, there simply must be more exposures than typically occur in a clinical trial program to detect it.
An adverse effect may be rare in clinical trials for a number of reasons. Perhaps it only occurs in genetically susceptible subgroup of patients. Perhaps it only occurs in patients with a comorbid medical condition (e.g., epilepsy) that was an exclusion criterion in the clinical trials of the drug. Perhaps it only occurs in the presence of a concomitant medication (i.e., a drug-drug interaction) that was not permitted during the clinical trials. Perhaps the adverse effect is time dependent and requires a longer exposure than typically occurs in clinical testing (for example, only 100 patients must be on an antidepressant for 1 year to meet the long-term exposure requirement for approval). Perhaps the effect is dose dependent and requires a higher dose than was used in the clinical trials.
A common question posed by clinicians about a new drug after it has been released is: "Can it be used in combination with drug X?" Another common question is: "Can the dose be pushed above the highest recommended dose in the package insert?" There are good reasons for both of these questions. Nevertheless, clinicians are often essentially doing research when they use a recently marketed drug in their patients -- since the way in which they use the drug and the patients they treat with it may differ substantially from how the drug was used in its clinical development trials.
The preceding discussion is not theoretical, but rather is relevant to the reason why thioridazine received a black box warning last summer, almost 40 years after it was first marketed. That development underscores the fact that acquiring knowledge about safety is an ongoing process in drug development that continues to evolve over time. Over the last decade, there has been a growing awareness that some drugs can cause a dose (i.e., concentration) dependent prolongation of intracardiac conduction as measured by a lengthening of the QT interval.23-25 While the first reports of sudden death on thioridazine occurred over 30 years ago,26 the label of thioridazine was not revised until data from the 054 study with ziprasidone documented a substantial prolongation of the QTc interval in patients treated with thioridazine. Thus, the changing labeling of thioridazine is an excellent illustration of the evolution of the requirements for proving both efficacy and safety that has occurred over the past 40 years. It will be interesting to see how our understanding of the efficacy and safety of recently released drugs will evolve over the next 40 years.
In the next column in this series, I will discuss the second wave of antipsychotics, the high-potency agents typified by haloperidol.
References
- Preskorn SH, Series of columns in this journal beginning with March 1998 and continuing from July 1999 through July 2000
- Oshiro Y, Sato S, Kurahashi N, et al, Novel antipsychotic agents with dopamine autoreceptor agonist properties: Pharmacology of 7-[4-(4phenyl-1-piperazinyl)butoxy]. J Med Chem 1998;41:658-67
- Lawler CP, Prioleau C, Lewis MM, et al, Interactions of the novel antipsychotic aripiprazole (OPC-24597) with dopamine-receptor subtypes. Neuropsychopharmacology 1999;20:612-27
- Lehmann HE, Ban TA, The history of the psychopharmacology of schiz ophrenia. Can J Psychiatry 1997;42:152-63
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Preskorn SH, Two in one: The venlafaxine story. J Pract Psychiatry Behav Health 1999;5:346-50
- Preskorn SH, Bupropion: What mechanism of action? Journal of Psychiatric Practice 2000;6:39-44
- Preskorn SH, Imipramine, Mirtazapine, and Nefazodone: Multiple targets. Journal of Psychiatric Practice 2000;6:97-102
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Laborit H, Huguenard P, Alluaume R, Un nouveau stabilisateur vegetatif, le 4560 RP. Presse Med. 1952; 60:206-208
- Delay J, Deniker Pl, Le traitement des psychoses par une m?thode neurolytique d?riv?e de I’hibermotherapie. Congr?s des m?decins alienistes et neurologistes de France. Luxembourg: July 1952: 497-502
- Kuhn R, The treatment of depressive states with G-22355 (imipramine hydrochloride). Am J Psychiatry. 1958;115: 459-464
- Preskorn SH, The stages of drug development and the human genome project: Drug discovery. Journal of Psychiatric Practice 2000;6:341-4
- Baldessarini R, Frankenburg FR, Clozapine: A novel antipsychotic agent. N Engl J Med 1991;324:746-54
- Meltzer HY, Pre-clinical pharmacology of atypical antipsychotic drugs: A selective review. Br J Psychiatry 1996;168:23-31
- Meltzer HY, Matsubara S, Lee LC, Classification of typical and atypical antipsychotic drugs on the basis of dopamine D 1,D2 and serotonin2 pKi values. J Pharmacol Exp Ther 1989;25:238-46
- Davis JM, Janicak PG, Lindon R, et al, Neuroleptics and psychotic disorders. In: Coyle JT, Enna SJ, eds. Neuroleptics: Neurochemical, behavioral and clinical perspectives. New York: Raven Press; 1983
- Thioridazine package insert. In: Physicians’ Desk Reference 2001, 55th edition. Montvale, NJ: Medical Economics Company; 2001
- Prim RF, Caffey EM, Klett CJ, Comparison of lithium carbonate and chlorpromazine in the treatment of mania. Arch Gen Psychiatry 1972;26:146-53
- Richelson E, Receptor pharmacology of neuroleptics: Relation to clinical effects. J Clin Psychiatry 1999;60(suppl 10):5-14
- Preskorn SH, Comparison of the tolerability of bupropion, fluoxetine, imipramine, nefazodone, paroxetine, sertraline and venlafaxine. J Clin Psychiatry. 1995;56(suppl 6):12-21
- Delpon E, Valenzuela C, Tamargo J, Blockade of cardiac potassium and other channels by antihistamines. Drug Saf 1999;21:11-8
- Tamargo J, Drug-induced torsades de pointes: From molecular biology to bedside. Jpn J Pharmacol 2000;83:1-19
- De Ponti F, Poluzzi E, Montanaro N, QT-interval prolongation by noncardiac drugs: Lessons to be learned from recent experience. Eur J Clin Pharmacol 2000;56:1-18
- Kelly HO, Fay JE, Lavery FG, Thioridazine hydrochloride (Mellaril): Its effect on the electrocardiogram and a report on two fatalities with electrographic abnormalities. Can Med Assoc J 1963;89:546-54
|



