|
|
| Drug Discovery in Psychiatry: Drilling Down on the Target of Interest |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice, July 2001, 267-272
|
The figures, in the exact sequence presented, form the essence of this column. Taken as a sequential whole, they illustrate the concept of "drilling down" on a target of interest. Looking at Figures 1 through 4, imagine that one is using a biochemical microscope to drill down into the physiology of interest to define novel sites of action (SOAs) for drug discovery.
Figure 1 is the most global illustration of a potential target, such as a novel G-protein coupled receptor. Figure 2 expands the postsynaptic receptor to show its relationship to other mechanisms within the postsynaptic cell. Figure 3 drills down even further on the postsynaptic receptor to show its conformation in two dimensions as it weaves through the plasmalemma of the postsynaptic neuron. Figure 4 magnifies the receptor further and illustrates the amino acid sequences that make up its transmembrane spanning segments. Thus, each figure expands on the figure preceding it and more precisely defines a potential target of interest for drug development. That is the essence of what is meant by "drilling down" on the target in drug discovery.
This column and these figures illustrate the implications of the human genome project for drug development in psychiatry and complement earlier columns on the evolution of antipsychotics and antidepressants.4,5 As pointed out in those columns, the modern era of drug discovery in psychiatry has progressed in epochs. The first epoch began in the late 1940s and early 1950s and was marked by a series of chance observations. The second epoch began in the late 1970s and extended through the early 1990s; it was marked by the use of receptor binding studies and other in vitro techniques to refine structure-activity relationships in order to synthesize compounds with a specific desired neural mechanism or mechanisms, while avoiding other, undesired mechanisms of action. The third epoch started in the early 1990s and has involved the use of techniques derived from molecular biology to discover completely new SOAs for drug development in psychiatry.
As mentioned earlier in this series on the human genome project and drug development,6 the SOA is the focus of the drug discovery phase of drug development. The SOA is the first variable in the following equation:
Effect = affinity for x drug x biological
site of action concentration variance
(Equation 1)
The SOA of a drug determines how the drug can alter physiology, which in turn determines whether and how that drug can help in a specific disease process. By binding to its SOA, a drug alters the physiological mechanisms regulated by that SOA. Parenthetically, the affinity of the drug for the SOA determines the concentration (i.e., the second variable in the equation) needed to engage (i.e., have agonist effects) or block (i.e., antagonize) the SOA to a clinically meaningful degree. For completeness, the third variable in equation 1 is the biological variance among humans that can shift the dose-response curve for a specific patient so that he or she is "sensitive" or "resistant" to the drug relative to the usual patient.7
The Second Epoch
Beginning with the second epoch in drug discovery, scientists could begin both dialing in and dialing out specific pharmacological effects by altering the structure-activity relationship so that the drug affected specific SOAs and avoided others. This is the reason that eight pharmacologically distinct classes of antidepressants are now available.4
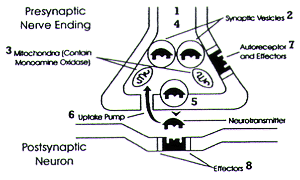
Reprinted with permission from Richelson 1994.1 | | Figure 1. Components of a synapse | | | | 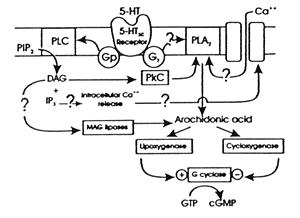
Reprinted with permission from Hartig et al. 19902 | | Figure 2. Linkage between a G-protein coupled receptor and its second messenger system | | | | 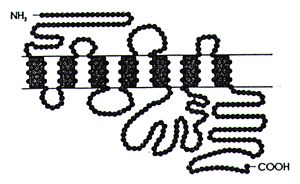
Reprinted with permission from Julius et al. 19983 Copyright 1988 American Association for the Advancement of Science. | | Figure 3. General structure of a G-protein coupled receptor for a biogenic amine neurotransmitter | |
Such drugs can be conceptualized as probes into brain function and thus into the pathophysiology that underlies psychopathology. Recall that Emil Kraepelin, MD, the father of modern psychiatry, anticipated this approach almost 100 years ago.8 The practicing psychiatrist can use this theoretical foundation and knowledge of the pharmacology of the drug and the patient's response to formulate a hypothesis of why the patient responded as he or she did and how to revise the patient's medication regimen, if clinically indicated. Recall that every "med check" is an assay based on equation 1 in which the goal is to determine whether the right SOA (variable 1) was engaged or disengaged to the right degree (variable 2) in the right patient (variable 3).
The NIH Synapse
Figure 1 has often been referred to as the "National Institute of Health (NIH) synapse." Virtually every psychiatrist and psychopharmacologist has seen this illustration or one like it innumerable times in lectures, books, and articles. Although this illustration may initially seem quite simple, from the point of view of drug discovery, the synapse it represents is rich in potential targets (i.e., SOAs). This figure alone shows 8 distinct SOAs that could be targets for specific drug development:
- the elements responsible for the synthesis of the neural transmitter
- the vesicle that concentrates and protects the neurotransmitter from elements in the cell's cytosol
- its degradatory enzyme, e.g., monoamine oxidase (MAO)
- the mechanisms that transport the vesicle from its site of formation to the terminal field
- the mechanisms that mediate fusion of the vesicle with the cell wall
- the uptake pump for the neurotransmitter
- its presynaptic receptor
- its postsynaptic receptor.
Depending on the pathophysiology of the disease being treated, one or more than one of these targets might mediate beneficial or adverse effects. For example, specific drugs can either improve or aggravate extrapyramidal motor function in patients with Parkinson's disease as a result of their effects on the specific SOAs shown in Figure 1. L-dopa remains the cornerstone of treatment for Parkinson's disease since it is the substrate which is converted into the neurotransmitter (SOA #1). Reserpine can aggravate Parkinson's disease by blocking the formation of vesicle (SOA #2). That results in the depletion of dopamine due to the inability to concentrate and protect it from degradation by MAO in the cell's cytosol. Amphetamines can cause extrapyramidal motor movements by causing the fusion of dopamine vesicles with the cell wall (SOA # 5) and also by blocking the dopamine uptake pump (SOA #6). These two actions are synergistic since the first increases the concentration of dopamine in the synaptic cleft and the second slows its clearance from the cleft. Bupropion and amantadine can also block the dopamine uptake pump (SOA #6) and thus can be used to improve extrapyramidal function in patients with idiopathic or neuroleptic-induced Parkinsonism. Neuroleptics or "typical" antipsychotics can cause extrapyramidal adverse effects such as Parkinsonism or aggravate the functional impairment of a patient with Parkinson's disease by blocking the postsynaptic dopamine-2 (D2) receptor (site #8). Conversely, a D2 agonist such as pergolide, pramipexole, or ropinirole could be used to treat Parkinson's disease and excessive doses could cause dyskinetic movements.9 Selegiline can increase the conservation of dopamine by inhibiting the B form of MAO (SOA #3), which preferentially degrades dopamine.

Reprinted with permission from Hartig et al. 19902 | | Figure 4. Comparison of the transmembrane spanning regions of the 5-HT2A, 5-HT2C, and 5-HT1A G-protein coupled receptors |
A Brief Detour
The preceding discussion is relevant to issues covered in an earlier series of columns on polypharmacy.10 For that reason, a brief detour will be taken from the main thrust of this column to illustrate how the principles underlying drug discovery are relevant to the use of more than one drug in combination.
As can readily be seen in Figure 1, some drugs such as L-dopa plus selegiline or L-dopa plus amantadine can be used together to enhance the treatment of a patient with Parkinson's disease. Other combinations, such as L-dopa plus haloperidol or haloperidol plus amantadine, could antagonize each other.
All of these combinations involve pharmacodynamic drug-drug interactions (DDIs). Some of these combinations are used as augmentation strategies. Others are used to treat adverse effects of the first drug (e.g., the addition of amantadine is a treatment for neuroleptic-induced extrapyramidal adverse effects such as Parkinsonism or acute dystonia). The combinations listed above all involve planned DDIs with the goal being to improve either efficacy or tolerability. In other instances, however, the DDI may be unintended and the outcome can be adverse. A column in my polypharmacy series described an example of an unintended DDI with an adverse outcome due to excessive antagonism of dopamine in the basal ganglia.11
Figure 1 also provides an illustration of how underlying biological variance can shift the dose-response curve of a drug. For example, haloperidol at sufficiently high doses (i.e., concentrations) can cause Parkinsonism in anyone. However, lower concentrations are needed in an individual with Parkinson's disease because they have less dopamine reserves to overcome the functional deficiency in D2 receptor stimulation produced by haloperidol blockade of the receptor. Another column in the polypharmacy series described such a case.12
The interested reader is referred to earlier columns for further discussion of the issues of polypharmacy,10 DDIs, and the conceptualization of how biological variance can shift the dose-response curve of specific drugs. These columns can be found in past issues of this journal or on the website.
Beyond the Postsynaptic Receptor
Having taken this brief detour, the column now returns to its focus on how scientists in drug discovery can drill down and refine their knowledge about targets of potential interest. Illustrations have been taken from papers published in the late 1980s and early 1990s to demonstrate that this approach is not new.
Figure 2 illustrates what lies beyond the postsynaptic receptor illustrated in Figure 1. Figure 2 is based on the serotonin or 5-hydroxytryptophan (5-HUC) receptor. This receptor belongs to the superfamily of G-protein coupled receptors (GPCR).13,14 This superfamily of regulatory proteins has provided a varied and fruitful array of targets for scientists in drug discovery. Many classes of psychiatric medications work, at least in part, through their actions on different members of this superfamily of receptors. In fact, that is why scientists are now searching the human genome for "orphan" GPCR, as discussed earlier in this series.15
As the name implies, the GPCR illustrated in Figure 2 is coupled to a G-protein, which is, in turn, coupled to a second messenger system. In the case of the 5-HT2C receptor, the second messenger system includes phospholipase C (PLC) and phospholipase A2 (PLA2), which in turn influence phosphokinase C (PkC). These second messenger systems regulate a biochemical cascade within the postsynaptic cell as illustrated in Figure 2. One result of that cascade is to modify the size of the calcium channel and thus the calcium current into the cell. That current in turn partly determines the cell's rate of output and its responsivity to afferent input.
Figure 2 thus presents a number of potential targets for scientists in drug discovery. For example, some investigational new drugs (IND), such as rolipram, have been developed with their target being the second messenger itself.16 To date, such drugs have not been successful, perhaps because the target is too fundamental to too many different physiological processes to be sufficiently well tolerated as a successful treatment, at least on its own. Nevertheless, Figure 2 illustrates how molecular biology techniques can be used to identify potential new targets for drug discovery. This phenomenon will only increase with further mining of the human genome.
Blowing Up the Receptor
Figure 3 expands the 5-HT2C receptor illustrated in Figure 2. Like all GPCRs, it has a nitrogen terminus outside the cell and a carboxyl terminus inside the cell. It weaves through the cell like a thread through fabric. It has seven transmembrane spanning segments and three intracytoplasmic loops. Changes in the conformation of the third loop are what initiate changes in the functional status of the second messenger Those conformational changes are the result of the interaction of the endogenous neurotransmitter with the receptor or the action of drugs which are either agonists, antagonists, or inverse agonists at the receptor.6
The circles in Figure 3 illustrate the amino acids that make up this regulatory protein. The sequence is determined by the nucleotide sequence in the gene that codes for this regulatory protein. This sequence also determines the three-dimensional conformation of the receptor and determines the docking regions where drugs can bind to the receptor and alter its functional status.
Blowing Up the Transmembrane Spanning Segments
Figure 4 illustrates the amino acid sequences that make up the first four spanning segments from the N-terminus end of three subtypes of 5-HT receptors: 1A, 2A, and 2C. These sequences were determined using molecular biological techniques available in the late 1980s. Such sequences can now be deduced from the nucleotide sequence of the gene coding for the receptor.
As illustrated in Figure 4, these three receptors demonstrate considerable sequence homology. This is why they all respond to serotonin as their endogenous neurotransmitter, and it is also the basis for their designation as 1A, 2A, and 2C. Receptor families, like the superfamily of cytochrome P450 enzymes and other regulatory proteins, are classified and grouped according to the degree of their sequence homology.17,18 The rationale for this approach comes from molecular biology. The more homologous the sequences of any two regulatory proteins are, the closer those proteins are likely to be evolutionarily.19 This is because the genes that code for them differ in the nucleotides that code for the different amino acids. Those changes in the nucleotide sequence resulted from mutations in a common ancestral gene that evolved into the current genes in the genome for a species. Those mutations also affect the function of the regulatory protein, including what neurotransmitter affects it.
These mutations paralleled the evolution of the brain. This is why different subtypes of receptors are incorporated to different degrees in different brain regions. This differential representation explains why a single neurotransmitter, such as serotonin, can be involved in so many different physiological processes, which in turn explains why drugs such as the selective serotonin reuptake inhibitors (SSRIs) can produce so many different actions. SSRIs are reasonably selective for one target, the serotonin uptake pump.20 However, that single action increases the availability of serotonin and hence its action on all of its various postsynaptic receptors.
The fact that different receptor subtypes for a single neurotransmitter share considerable sequence homology and hence conformational structure is also the reason why a drug may affect several different members of the same receptor family, even though the desired effect of the drug is mediated by its action on only one of those receptors. That is the power of the information illustrated in Figure 4. For the purposes of drug discovery, knowledge of the amino acid sequence for a regulatory protein like a specific 5-HT receptor is analogous to knowing the tumbler system of a lock one wants to pick. The sequence determines the conformation of the receptor and determines where and how the drug can alter that conformation. Thus, knowledge of the information illustrated in Figure 4 permits scientists in drug discovery to refine structure-activity relationships to guide the rational development of a new chemical entity that is selective for only one receptor subtype within a family.6
In the earlier series on antidepressants, the concept of selectivity was explained using a simple figure showing three different drugs. one with a 3-fold separation in its ability to bind to receptor X versus Y, a second drug with a 10-fold separation in its affinity for these same two receptors, and a third drug with a 100-fold separation.4 Using the knowledge in Figure 4, drug discovery scientists can pick their targets of interest and dial in the effect of their drug on a specific SOA or SOAs depending on whether they want a drug with a "selective" pharmacology (e.g., sertraline) or with a sequential effect on two different targets (e. g., venlafaxine) or one with affinities for a multitude of targets (e.g., amitriptyline).4 This was the same theme discussed in the column on the evolution of antipsychotic medications.5
Creating Receptor Variants
Changes in the amino acid sequence of a regulator protein do not necessarily change either its conformation or its function, or the binding affinity of a drug for it. Some amino acid changes are silent. This is the reason not all mutations are functionally meaningful. In the 1980s and early 1990s, drug discovery scientists had to rely on the natural existence of receptor subtypes to develop their knowledge of the important amino acid sequences that conveyed important functional and binding differences. That approach is necessarily limited by the number of receptor subtypes that exist. Using molecular biology techniques, drug discovery scientists can now swap a given amino acid in the sequence of regulatory protein or a nucleotide in the gene and see how that alters either the function or conformation of the regulatory protein. The resulting information can be used to test and further refine structure-activity relationships for drug discovery.
At the same time, the human genome can be mined for previously unknown regulatory proteins that may be useful targets for drug discovery.21 When such targets are identified, then the process illustrated in Figures 1 through 4 can be used to develop lead compounds. As discussed in earlier columns,6 the brain is already known to have a large number of targets that have not yet been fully explored in terms of drug development. Those numbers will increase with further mining of the human genome.
The drug leads that will result from these efforts hold the promise of quite novel approaches to the treatment of neuropsychiatric diseases as well as probes into the pathophysiology underlying these illnesses, as Kraepelin foresaw almost 100 years ago. However, this promise brings with it costs and responsibilities.
As the pharmacology of psychiatric medications becomes more focused on and limited to neural mechanisms of action, their adverse effects will be on the brain rather than on peripheral systems. These adverse effects will likely present as behavioral problems (e.g., confusion) rather than as peripheral toxicity (e.g., dry mouth, cardiac arrhythmias). For this reason, psychiatrists and others who prescribe psychiatric medications will need to be accomplished behavioral pharmacologists sensitive to all of the possible effects of the drug. By increasing the numbers and types of psychiatric medications available, this approach will further increase the potential for even more polypharmacy than is already practiced today Hopefully, increased knowledge of the physiology of the brain will permit more informed rationale for the use of polypharmacy in the treatment of patients with psychiatric illnesses than is currently possible.
The expanding array of potential targets also presents significant challenges for psychiatric drug discovery and development. The number of potential targets is so large that it will not be cost effective to pursue every one of them. Instead, targets that are likely to be clinically useful will have to be carefully selected. That will require studies aimed at establishing some idea about the role the mechanism subsumes in the human brain. Once that has been accomplished and looks promising, there will be an increased need to better characterize the behavioral pharmacology of the new candidate agent in man rather than in animals, given that such drugs will be finely targeted to specific mechanisms in the human brain. This will involve more use of "bridging" studies in volunteers who are mildly symptomatic with the target illness in question.6 This is because the pathophysiology of the disease being treated (variable 3) is likely to influence the response to the drug (equation 1). Ironically, most drugs today are developed based on tests using normal subprimate brains or brain tissue. Yet these drugs are given to patients who presumably have abnormal brain function as a result of their illness.
All of these issues will be the focus of subsequent columns in this series.
References
- Richelson E, Pharmacology of antidepressants: Characteristics of the ideal drug. Mayo Clin Proc. 1994;69:1069-1081
- Hartig PR, Hoffman BJ, Kaufman MJ, et al, The 5-HT1C receptor. Ann NY Acad Sci 1990;600:149-66
- Julius D, MacDermott AB, Axel R, et al, Molecular characterization of a functional cDNA encoding the serotonin 1c receptor. Science 1988; 241:558-64
- Preskorn SH, Understanding the basics of antidepressant pharmacotherapy. (collection of columns published in the Journal of Psychiatric Practice) Accessed June 25, 2001
- Preskorn SH, Antipsychotic drug development in the pre-human genome era: A full circle. Journal of Psychiatric Practice 2001;7:209-213
- Preskorn SH, Modern drug development and the Human Genome Project. (collection of 4 columns published in the Journal of Psychiatric Practice). Accessed June 25, 2001
- Preskorn SH, The rationale for and clinical implications of dose-response curvese project. (collection of columns published in J Pract Psychiatry Behav Health).
- Kraepelin E, Dementia praecox and paraphrenia. Translated by Barclay RM. Robertson GM, ed. Huntington. NY Krieger; 1971
- Tuite P, Ebbitt B, Dopamine agonists. Semin Neurol 2001;21:9-14
- Preskorn SH, The polypharmacology section. (collection of columns published in the Journal of Psychiatric Practice). Accessed July 10, 2002
- Preskorn SH, I don’t see ’em. J Prac Psych Behav Hlth. 1997; 3:302-307
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Brauner-Osborne H, Jensen AA, Sheppard PO, et al, Cloning and characterization of a human orphan family C G-protein coupled receptor GPACSD. Biachim Biophys Acts 2001;1518:237-48
- Ballesteroa JA, Shi L, Javitch JA, Structural mimicry in G protein-coupled receptors: Implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol 2001;60:1-19
- Preskorn SH, The human genome project and drug discovery in psychiatry: Identifying novel targets. Journal of Psychiatric Practice 2001;7:133-7
- Ye Y, Jackson K, O’Donnell JM, Effects of repeated antidepressant treatment of type 4A phosphodiesterase (PDE4A) in rat brain. J Neurochem 2000;74:1257-62
- Hoyer D, Clark DE, Fozard JR, et al, International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacol Rev. 1994;46:157-203
- Nelson DR, Kamataki T, Waxman DJ, et al, The P450 superfamily: update on new sequences, gene mapping, accession numbers, early trivial names of enzymes and nomenclature. DNA Cell Biol. 1993;12:1-51
- Wittenberger T, Schaller HC, Hellebrand S, An expressed sequence tag (EST) data mining strategy succeeds the discovery of new G-protein coupled receptors. J Mol Biol 2001;307:799-813
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Landro JA, Taylor IC, Stirtan WG, et al, HTS in the new millennium: The role of pharmacology and flexibility. J Pharmacol Toxicol Methods 2000;44:273-89
|



