|
|
| The Relative Adverse Effect Profile of Non-SSRI Antidepressants: Relationship to In Vitro Pharmacology |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice, July 2000, 218-223
|
This column is the seventh in a series devoted to a discussion of the relationship between the in vitro binding affinity and the clinical pharmacology of the various classes of antidepressants. In my last column, I reviewed this issue as it relates to the adverse effect profiles of the five marketed selective serotonin reuptake inhibitors (SSRIs).1 In this column, I will extend the discussion to what I call (for lack of a better term) the non-SSRI antidepressants: bupropion, imipramine, mirtazapine, nefazodone, and venlafaxine.2 Parenthetically, in this discussion, imipramine serves as the prototype for a tertiary amine tricyclic antidepressant.
Figure 1 shows the relative binding affinity of these different antidepressants for specific neural targets of clinical significance (e.g., uptake pumps, neuroreceptors). A similar figure in my last column1 illustrated the relative binding affinity of the five SSRIs for the same specific neural targets. The relative binding affinities of the non-SSRI antidepressants differ substantially from the SSRIs as a group and from each other. Whereas each of the SSRIs has at least one order of magnitude separation between its binding affinity for the serotonin uptake pump and its affinity for its next most potent potential site of action, that is not true for the non-SSRI antidepressants.7
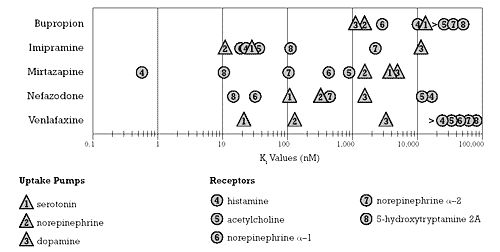 | Figure 1 - Relative Potency for Different Sites of Action of non-SSRI antidepressants
- Based on data from Bolden-Watson C, Richelson E, 1993,3, Cusack B, et al. 1994,4, and deBoer et al. 1988,5 19956 |
Of the non-SSRI antidepressants, venlafaxine most closely approximates the binding affinity profile of an SSRI. There is wide separation between its binding affinity for the serotonin and norepinephrine uptake pumps and other neural targets. Nevertheless, venlafaxine is not "selective" like the SSRIs--this is because there is less than a 5-fold separation between venlafaxine’s ability to block the serotonin uptake pump and its ability to block the norepinephrine uptake pump.7
| Table 1. Placebo-adjusted, dose-dependent adverse effects of paroxetine* | Adverse
effect | Paroxetine dose (mg/day) | 10
(n=102) | 20
(n=104) | 30
(n=101) | 40
(n=102) | | Nausea | 1.0 | 13.2 | 21.0 | 22.6 | | Sweating | -1.0 | 4.7 | 6.9 | 9.8 | | Dizziness | 3.0 | 2.8 | 5.0 | 8.8 | | Somnolence | 4.9 | 10.5 | 13.0 | 13.8 | Abnormal
Ejaculation | 5.8 | 6.5 | 10.6 | 13.0 | | Constipation | -1.0 | 1.8 | 4.0 | 6.8 | | | * Reprinted with permission from Janicak P, Davis JM, Preskorn SH, Ayd F Jr. Principles and practice of psychopharmacotherapy. Baltimore: Williams & Wilkins; 19978 | | | Table 2. Placebo-adjusted, dose-dependent adverse effects of venlafaxine* | Adverse
effect | Venlafaxine dose (mg/day) | 75
(n=89) | 225
(n=89) | 375
(n=88) | | Nausea | 18.5 | 24.1 | 43.9 | | Sweating | 1.3 | 7.0 | 13.9 | | Dizziness | 14.8 | 18.2 | 19.6 | | Somnolence | 12.6 | 13.7 | 21.8 | | Tremor | 1.1 | 2.2 | 10.2 | | Hypertension | 0 | 1.1 | 3.4 | | Yawning | 4.5 | 5.6 | 8.0 | | Chills | 1.1 | 4.5 | 5.7 | | | * Reprinted with permission from Janicak P, Davis JM, Preskorn SH, Ayd F Jr. Principles and practice of psychopharmacotherapy. Baltimore: Williams & Wilkins; 19978 | |
The dose-dependent adverse effects of paroxetine and the immediate release (IR) version of venlafaxine are listed in Tables 1 and 2, respectively. I am using paroxetine as a representative SSRI to contrast that selective class of drugs with venlafaxine, which sequentially engages two different mechanisms over its clinically relevant dosing range.9 I chose paroxetine because data from a fixed dose study are contained in its package insert illustrating the dose-dependent nature of some of its adverse effects. I show data for the IR rather than the extended release (ER) formulation of venlafaxine for the same reason (i.e., data on the adverse effects as a function of dose are given in the package insert for the IR but not the ER formulation).
In these two tables, I list the adverse effects that are clearly dose-dependent using the criteria that the placebo-subtracted incidence rate (i.e., rate on each dose minus the rate in the parallel placebo group) was higher for each higher dose. Parenthetically, the data in Tables 1 and 2 show that classic dose-dependent response curves can be found in clinical trials with psychiatric medications.
The adverse effects of nausea, somnolence, and abnormal ejaculation listed in Table 1 for paroxetine are generally recognized as typical of the SSRIs. These effects also occur with venlafaxine, consistent with its ability to block serotonin uptake. While sweating and dizziness might be thought by some to be less typical of the SSRIs, these adverse effects occur with a placebo-subtracted incidence rate of 4% or higher for three out of the five SSRIs.1 They also occur with venlafaxine in a dose-dependent fashion. This is not true of constipation, a dose-dependent adverse effect of paroxetine that could be consistent with its weak effect as a muscarinic cholinergic blocker.1 If that is the mechanism, the weakness of this effect is evident from the fact that the placebo-subtracted incidence rate is only 6.8% at 40 mg/day of paroxetine. Contrast that incidence with the rate on imipramine (Tables 3 and 4).
In contrast to paroxetine, venlafaxine causes a dose-dependent increase in blood pressure (Table 2). This effect is most likely due to the norepinephrine uptake inhibition produced by higher doses of venlafaxine. Consistent with this explanation, venlafaxine does not cause a detectable incidence of hypertension at 75 mg/day. That dose has also been shown not to blunt the tyramine pressor effect, which is an in vivo surrogate measure for norepinephrine uptake inhibition.9
Venlafaxine also causes a dose-dependent increase in tremors. That effect could be due to norepinephrine uptake inhibition. However, this adverse effect, as reported in clinical trials, is difficult to interpret with confidence since it also occurs with an incidence of more than 4% above placebo with four of the five SSRIs.1 This finding may reflect the fact that no neuron is an island (i.e., the neural systems in the brain are interconnected). Thus, the SSRIs may produce tremors through a serotonin-mediated effect on either the norepinephrine or dopamine systems.13
The point of Tables 1 and 2 is two-fold. First, some dose-dependent effects can be demonstrated in clinical trials with psychiatric medications. Ironically, they tend to be adverse as opposed to beneficial effects, in part because of the signal-to-noise problems inherent with efficacy measures.14 Second, we can often understand and even anticipate these adverse effects through a knowledge of the in vitro pharmacology of the drug coupled with a knowledge of the concentration of the drug achieved under clinically relevant dosing conditions.
Thus, the qualitative and quantitative changes in adverse effects as a function of different doses, shown in Tables 1 and 2, are readily understood from the equation I have frequently cited in these columns:
(Equation 1)
Effect = affinity for x drug x biological
site of action level variance
The highest affinity of venlafaxine is for the serotonin uptake pump. This is why venlafaxine, at its lowest effective antidepressant dose, causes serotonin-mediated adverse effects just like paroxetine. However, the adverse effects of venlafaxine change qualitatively at higher doses because it is capable of producing progressively greater blockade of norepinephrine uptake as the dose increases over its clinically relevant dosing range.9 Thus, the 5-fold in vitro difference in venlafaxine’s binding affinity for these two pumps has clinically meaningful consequences.
In contrast, the adverse effect profile of paroxetine stays generally the same qualitatively and increases only quantitatively over its clinically relevant dosing range. In other words, paroxetine’s adverse effect profile remains the same at higher doses, but these same adverse effects occur either in more patients (i.e., at a greater frequency) or with more severity. This is because, at higher doses, the SSRIs are simply blocking serotonin uptake to a greater degree.14 Paroxetine, like venlafaxine, is able to inhibit norepinephrine uptake in vitro; however, it is over 100 times more potent at blocking the serotonin than the norepinephrine uptake pump.1 Thus, the dose of paroxetine needed to block the norepinephrine uptake pump would be more than 100-fold higher than that needed to block the serotonin uptake pump--this is why paroxetine, like the other SSRIs, is called "selective." This is also why it would not be prudent to attempt to use paroxetine clinically to block norepinephrine uptake even though it would theoretically be possible. Even if cost were not an issue, most patients probably would not tolerate the degree of serotonin agonism that would result from a dose of paroxetine high enough to block norepinephrine uptake. Instead, the prescriber can accomplish the goal by either adding a selective norepinephrine uptake inhibitor such as desipramine to the treatment regimen15 or switching to a drug like venlafaxine, which is capable of blocking both targets over its clinically relevant dosing range.9 Parenthetically, the dose-dependent effect of venlafaxine on the serotonin and norepinephrine uptake pumps is also a heuristic explanation for why it has an ascending dose-antidepressant response curve in contrast to the flat dose- antidepressant response curve seen with the SSRIs. The pharmacological advantage of using two drugs in combination (i.e., an SSRI and a NSRI) is that the degree of blockade of one pump can be varied independently of the other. Venlafaxine is essentially a fixed combination product in which the uptake inhibition of serotonin will always exceed that of norepinephrine.
As can readily be seen in Figure 1, the non-SSRI antidepressants as a group are defined by what they are not rather than by what they are. Beyond being antidepressants, the only other feature that they share is that they are not pharmacologically selective in contrast to the SSRIs. However, the in vitro pharmacology of each of these drugs differs substantially. The question is whether this difference in in vitro pharmacology translates into meaningful differences in their clinical pharmacology. One way to assess for differences in the clinical pharmacology of these medications is to examine their relative risk of causing specific adverse effects.
Table 3. Comparison of the placebo-subtracted incidence rate (%) of frequent adverse effects for bupro-pion,
imipramine, mirtazapine, nefazodone, and venlafaxine* , 10–12 | Adverse
effect | Bupropion | Imipramine | Mirtazapine | Nefazodone | Venlafaxine-IR | Venlafaxine-ER | (n=323,
n=185)a | (n=367,
n=672)a | (n=453,
n=361)a | (n=393,
n=394)a | (n=1033,
n=609)a | (n=357,
n=285)a | | Anorexia | -0.1 | NA | NA | NA | 9 | 4 | | Confusion b | 2.8 | NA | 2 | 9 | 1 | 2 | | Constipation | 8.7 | 17.4 | 6 | 6 | 8 | 3 | | Diarrhea c | -1.8 | -2.7 | < placebo | 1 | 1 | < placebo | | Dizziness d | 6.8 | 22.7 | 4 | 23 | 12 | 11 | | Drowsiness e | 0.3 | 12 | 36 | 11 | 14 | 9 | | Dry mouth | 9.2 | 47.1 | 10 | 12 | 11 | 6 | | Dyspepsia | 0.9 | NA | < placebo | 2 | 1 | < placebo | | Fatigue f | -3.6 | 7.6 | 3 | 7 | 6 | 1 | | Flatulence | NA | NA | < placebo | NA | 1 | 1 | | Frequent micturition | 0.3 | NA | 1 | 1 | 1 | NA | | Headache | 3.5 | -8.7 | < placebo | 3 | 1 | < placebo | | Increased appetite | NA | NA | 15 | NA | 1 | 2 | | Insomnia | 5.3 | 0.4 | < placebo | 2 | 8 | 6 | | Nausea g | 4 | 1.3 | < placebo | 11 | 26 | 21 | | Nervousness | 13.9 | 3.6 | < placebo | NA | 12 | 7 | | Palpitations i | 4.7 | NA | < placebo | NA | 2 | < placebo | | Paresthesia j | 0.8 | NA | NA | 2 | 1 | NA | | Rash k | 3.7 | NA | NA | 2 | 1 | NA | | Respiratory l | -2.5 | -2.3 | 3 | 9 | NA | 1 | | Sweating | 7.7 | 11.2 | < placebo | NA | 9 | 11 | | Tremors | 13.5 | 10 | 1 | 1 | 4 | 3 | | Urinary retention m | -0.3 | 4 | NA | 1 | 2 | NA | | Vision disturbances | 4.3 | 5.4 | < placebo | 12 | 4 | 4 | | Weight gain | NA | NA | 10 | NA | NA | NA | | * The adverse effect data presented above come from product labeling as opposed to head-to-head trials. Such data may not necessarily reflect the actual rate of these adverse effects in clinical practice or the actual differences between these vari-ous drugs.
a The first value is the number of patients on that medication, while the second represents those treated in the parallel place-bo group.
b Includes decreased concentration, memory impairment, aban-doned thinking concentration
c Includes gastroenteritis
d Includes lightheadedness, postural hypotension, and hypotension
e Includes somnolence, sedation, and drugged feeling
f Includes asthenia, myasthenia, and psychomotor retardation
g Includes vomiting
h Includes anxiety, agitation, hostility, akathisia, and central nervous system stimulation
i Includes tachycardia and arrhythmias
j Includes sensation disturbances and hypesthesia
k Includes pruritus
l Includes respiratory disorder, upper respiratory infection, flu, dyspnea, pharyngitis, sinus congestion, oropharynx disorder, fever, and chill
m Includes micturition disorder, difficulty with micturition, and urinary hesitancy |
To make this comparison, I examined the incidence of adverse effects on each of the non-SSRI antidepressants in comparison to the parallel placebo condition in the double-blind clinical trials used for the approval of these drugs by the Food and Drug Administration. Based on these data, I calculated the attributable incidence rate of adverse effects for each of these antidepressants by taking the incidence of a given adverse effect in the group treated with a non-SSRI antidepressant minus the incidence of the same adverse effect in the parallel group treated with placebo (Table 3). The attributed incidence is the incidence that is specifically due to the drug as opposed to being due to other factors such as the underlying illness (e.g., major depression). The attributed risk then is a clinically useful measure of how many patients are likely to experience the adverse effect when taking the drug compared to an identical group of patients exposed to placebo rather than the drug.
The same results are also shown in Table 4, perhaps in a more user-friendly way. In this table, I show the attributable incidence of given adverse effects in frequency categories such as > 7.5% but < 10%. Using this table, the reader can determine at a glance the relative likelihood of a specific adverse effect on a given non-SSRI antidepressant relative to the other non-SSRI antidepressants.
| Table 4. The most likely adverse effects on specific non-SSRIs above and beyond the parallel placebo condition (percentage on drug minus percentage on placebo based on registration studies)* | | Antidepressant | > 7.5% | > 10% | > 15% | > 20% | > 25% | > 30% | > 35% | > 40% | > 45% | | Bupropion | dry mouth
constipation
sweating | nervousness
tremors | -- | -- | -- | -- | -- | -- | -- | | Imipramine | fatigue | drowsiness
tremors
sweating | constipation | dizziness | -- | -- | -- | | dry mouth | | Mirtazapine | -- | weight gain
dry mouth | increased
appetite | -- | -- | -- | drowsiness | -- | -- | | Nefazodone | confusion
respiratory | drowsiness
vision disturbance
nausea
dry mouth | -- | dizziness | -- | -- | -- | -- | -- | | Venlafaxine-IR | insomnia
anorexia
constipation
sweating | drowsiness
nervousness
dizziness
dry mouth | | -- | nausea | -- | -- | -- | -- | | Venlafaxine-ER | drowsiness | sweating
dizziness | -- | nausea | -- | -- | -- | -- | -- | | | *Data in this table are based on the information presented in Table 3. The adverse effect data presented above come from product label-ing as opposed to head-to-head trials. Such data may not necessarily reflect the actual rate of these adverse effects in clinical practice or the actual differences between these various drugs. |
Recall that I mentioned in my last column1 that the data on adverse effects given in the package insert (i.e., the basis for the numbers shown in Tables 3 and 4) are a function of the average dose used in the clinical trials pro-gram that led to the drug’s approval. An illustration will make it easier to understand the importance of this statement. Assume that we are comparing the incidence of adverse effects reported during the registration studies for two SSRIs (A and B). Let us say that both have the same recommended dosing range (20-60 mg/day). Let us further specify that 20 mg/day was eventually found to be the optimal dose for both of these SSRIs and that, at this dose, they produce exactly the same efficacy, a finding that was, however, not known during the entire development process for both the SSRIs. Let us further say that the company that developed SSRI A used aggressive dosing, so that the average dose over the course of its clinical trial program was 50 mg/day, whereas the company that developed SSRI B used a conservative dosing strategy, so that the average dose over its clinical trial program was 25 mg/day. The relative incidence of adverse effects, as given in the package insert, will be higher on A than on B because the incidence and the magnitude of the effect is a function of the concentration achieved at the site of action. However, SSRIs A and B may actually have a virtually identical incidence of adverse effects if compared at their comparably effective dose of 20 mg/day. To understand the potential difference in the incidence rate, look at Table 1 and assume that SSRI A is paroxetine, 40 mg/day, and SSRI B is paroxetine, 20 mg/day. SSRI B would look to be much better tolerated than SSRI A. This dosing issue would apply to all the data in Tables 3 and 4 because these data are based on the entire database developed during the clinical trials of these medications. For this reason, these data are only a guide to the relative rate of adverse effects; small differences are of dubious significance and even large differences must be interpreted cautiously. Recall that drug level is one of the three variables in Equation 1 that determines the nature and magnitude of a drug’s effect. Drug concentration, in turn, is a function of the dose given to the patient relative to his or her ability to clear the drug as expressed in the following equation:
(Equation 2)
Concentration = Dosing rate / Clearance
With this caveat in mind, I believe the data in Tables 3 and 4 are consistent with the conclusion that the differences in the in vitro pharmacology of the non-SSRI anti-depressants are clinically relevant in terms of their human clinical pharmacology under their usual antidepressant dosing conditions. Whereas the SSRIs, as I discussed in my last column, produce virtually the same adverse effects, the adverse effect profiles of the non-SSRI antidepressants differ substantially from each other in a manner that is fully consistent with the differences in their in vitro binding affinities.
Again, in contrast to the data I presented in my last column, the differences that exist in Tables 3 and 4 are qualitative (i.e., differences in the nature of the effects) rather than simply quantitative (i.e., differences in the frequency or severity of the effects). That is readily apparent when one examines the most frequent adverse effects of these different antidepressants, starting from the right side of Table 4 (i.e., the most frequent effects) and moving to the left side of the table (i.e., the least common effects). Thus, dry mouth occurs in more than 45% of patients on imipramine, which is consistent with its ability to bind to and block muscarinic acetylcholine receptors. Drowsiness occurs in more than 35% of patients on mirtazapine, consistent with its most potent action, the blockade of histamine-1 receptors. Consistent with its ability to block the uptake of serotonin, the most common adverse effect of venlafaxine (in both the IR and the ER formulations) is nausea, just as with the SSRIs.
Sometimes there may be more than one reason why a drug could cause an adverse effect. Take, for example, the dizziness reported on nefazodone. This effect could be due to the ability of nefazodone to block the alpha-1 adrenergic receptor. However, it may also be due to indirect stimulation of the serotonin-1A (more often termed the 5-hydrox-ytryptophan [5-HT] 1A receptor) as a result of 5-HT2A blockade. This latter hypothesis is based two observations. First, the dizziness that occurs with nefazodone is qualitatively different from that seen with alpha-1 adrenergic blockade and is generally not accompanied by an orthostatic drop in blood pressure as occurs with a drug such as imipramine. Second, the dizziness reported with nefazodone is similar to that caused by azapirones, such as buspirone, which are 5HT1A agonists.
An adverse effect may also be due to more than one mechanism of action. While the dry mouth on imipramine is most likely due to its ability to block muscarinic acetylcholine receptors, that mechanism is not likely to be responsible for the dry mouth that occurs with mirtazapine and venlafaxine. Both of these drugs are so weak as blockers of the muscarinic acetylcholine receptors that this action is unlikely to be clinically relevant at usual doses. In contrast, both drugs can potentiate the effects of norepinephrine, although by different mechanisms. In the case of mirtazapine, the mechanism is likely alpha-2 adrenergic receptor blockade, which can increase norepinephrine release. In the case of venlafaxine, the most likely mechanism is norepinephrine uptake inhibition. Thus, dry mouth on these two drugs most likely occurs as a result of norepinephrine agonism rather than acetylcholine antagonism.
The point of this column, like the others before it, is that we can anticipate the adverse effects of a drug based on its in vitro pharmacology, realizing that we must take drug concentration into account. A drug will not exert an action if it does not achieve a concentration that allows it to occupy a target to a critical degree under clinically relevant dosing conditions. When the mechanism is uncertain, further data can help to determine what is happening. Such data can sometimes be obtained clinically (e.g., by measuring orthostatic blood pressure). In other situations, different tests (e.g., measuring the inhibition of serotonin uptake in platelets or the pressor response to intravenous administration of tyramine as a measure of the degree of norepinephrine uptake inhibition) can be done.9
As can be readily seen from this column and those that have preceded it in this series, the same mechanism that accounts for a drug’s beneficial effects is often also responsible for its adverse effects. This is particularly true for drugs that have been designed to be highly selective in terms of their mechanisms of action, such as the SSRIs and venlafaxine.
References
- Preskorn SH, The adverse effect profiles of the selective serotonin reuptake inhibitors: Relationship to in vitro pharmacology. Journal of Psychiatric Practice 2000;6:153-7
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Bolden-Watson C, Richelson E, Blockade by newly-developed antidepressants of biogenic amine uptake into rat brain synaptosomes. Life Sci. 1993;52:1023-1029
- Cusack B, Nelson A, Richelson E, Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559-565
- de Boer TH, Maura G, Raiteri M, de Vos CJ, Wieringa J, Pinder RM, Neurochemical and autonomic pharmacological profiles of the 6-aza-analogue of mianserin, Org 3770 and its enantiomers. Neuropharmacology. 1988;27:399-408
- DeBoer TH, Ruigt GSF, Berendsen HHG, The alpha 2 - selective adrenoceptor antagonist Org 3770 (mirtazapine, Remeron) enhances noradrenergic and serotonergic transmission. Human Psychopharmacology 1995;10:S107-S118
- Preskorn SH, De-spinning in vitro data. J Pract Psychiatry Behav Health 1999;5:283-7
- Janicak PG, Davis JM, Preskorn SH, Ayd FJ Jr, Principles and Practice of Psychopharmacotherapy. 2nd ed. Baltimore, Md: Lippincott, Williams & Wilkins; 1997
- Harvey AT, Rudolph R, Preskorn SH, Evidence of the dual mechanism of action of venlafaxine in healthy male volunteers. Arch Gen Psychiatry, 2000;57:503-9
- Preskorn SH, Comparison of the tolerability of bupropion, fluoxetine, imipramine, nefazodone, paroxetine, sertraline and venlafaxine. J Clin Psychiatry. 1995;56(suppl 6):12-21
- Physicians’ Desk Reference, 53rd Edition. Prescribing information for mirtazapine. Montvale, NJ: Medical Economics Co.; 1999:2147-9
- Physicians’ Desk Reference, 53rd Edition. Prescribing information for venlafaxine hydrochloride. Montvale, NJ: Medical Economics Co.; 1999:3298-3302
- Preskorn SH, I don’t see ’em. J Prac Psych Behav Hlth. 1997; 3:302-307
- Preskorn SH, Finding the signal through the noise: The use of surrogate markers. J Pract Psychiatry Behav Health 1999;5:104-109
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
|



