|
|
| Reproducibility of the In Vivo Effect of the Selective Serotonin Reuptake Inhibitors on the In Vivo Function of Cytochrome P450 2D6: An update (Part II) |
|
|
SHELDON H. PRESKORN, MD
|
|
Journal of Psychiatric Practice, May 2003, 228-236
|
This column is the second in a two-part series explaining how the effects of drugs on human drug metabolizing enzymes such as cytochrome P450 (CYP) 2D6 are studied and quantified. A considerable amount of research has focused on the fact that some drugs-- including some antidepressants--are capable of producing substantial inhibition of one or more human drug metabolizing enzymes under usual dosing conditions. This work has demonstrated that drugs within the same therapeutic class and even the same pharmacodynamic class can have substantial and clinically meaningful differences in their effects on such enzymes and that such differences can be important in choosing among members of a therapeutic class.
The importance of such enzyme inhibition has been discussed in earlier columns.1-11 One column pointed out that screening for such unintended effects on human drug metabolizing enzymes is now part of the earliest testing of new candidate drugs and that the results of such tests are used to make "Go, No Go" decisions about which drugs to take forward into human testing.12 Another column pointed out that substantial unintended inhibition of such enzymes has resulted in the withdrawal of a number of drugs from the U.S. market over the past decade.7 These withdrawals occurred when it was recognized that the co-administration of these drugs sets the stage for serious and even fatal drug-drug interactions when the inhibitor drug ("perpetrator") is used in combination with a substrate drug ("victim") that has a narrow therapeutic index and is predominantly metabolized by the inhibited enzyme.
The goal of this two-part series is to explain how such effects are measured in living human beings under usual dosing conditions of the inhibitor. The impetus for this column was frequent questions from clinicians about how these studies are done, how the results are measured, and the reproducibility of the reported differences among drugs within a therapeutic class.
How can drugs be meaningfully classified?
According to Websters New Collegiate Dictionary, a class is "a group, set, or kind sharing common attributes," "a division...based on grade or quality."
Drugs can be classified in many ways. They can be classified in terms of chemical structure, which is usually a turnoff to physicians because it tells most physicians little about what the drug does. Drugs can be classified according to clinical use (i.e., therapeutic class) which is quite meaningful to clinicians, even if it may be relatively uninformative from a biological or pharmacological perspective. For example, knowing that a drug is an antidepressant tells one little about the pharmacology of the drug, given that there are currently eight classes of antidepressants based on the putative mechanism underlying their antidepressant efficacy.13,14 Drugs can also be classified according to their principal mechanism of action (i.e., pharmacodynamics) or according to their pharmacokinetics (e.g., by what enzymes they are metabolized and how they are eliminated).
For example, the selective serotonin reuptake inhibitors (SSRIs) belong to many different classes when viewed from these various perspectives: structure, therapeutic use, pharmacodynamics, and pharmacokinetics. The SSRIs belong to the superfamily of antidepressants. The SSRIs were the first class of psychiatric medications that were rationally developed to have a single central mechanism of action, serotonin uptake inhibition, using in vitro receptor-binding technology.12,15 Thus, SSRIs can be defined as belonging both to the class of antidepressants (a therapeutic class) and to the subclass of serotonin uptake inhibitors (a pharmacodynamically defined class).
However, while the six marketed SSRIs are alike in these ways, they belong to different classes when considered from the perspective of their unintended and undesired ability to inhibit specific CYP enzymes, which is another clinically important mechanism-based class.
| Table 1. Summary of the in vivo effect of specific doses of SSRIs on the major human drug metabolizing cytochrome P450 enzymes* | | | 1A2 | 2D6 | 2C9/10 | 2C19 | 3A3/4 | | Citalopram | • | ++ | • | • | • | | Escitalopram | • | ++ | • | • | • | | Fluoxetine | • | +++ | +++ | ++ | + | | Fluvoxamine† | | • | +++ | +++ | ++ | | Paroxetine | • | +++ | • | • | • | | Sertraline | • | + | • | • | • |
| • No or minimal effect (< 20%)
+ Mild effect (20%-50%
++ Moderate effect (50%-150%)
+++ Substantial effect (> 150%)
Sources: Preskorn 199615 and 199914; Harvey and Preskorn 199616,17 and Madsen et al. 200118 Copyright Preskorn 2003
*The information in the table is based on the results of formal in vivo studies in humans. The ratings are based on the following doses because the effects are dose dependent and the studies were principally conducted at these doses: 40 mg/day of citalopram; 20 mg/day of escitalopram, fluoxetine, and paroxetine; 150 mg/day of fluvoxamine; and 100 mg/day of sertraline, with some studies testing doses as high as 200 mg/day.
†This table has been updated to reflect the results of a recent vivo human study showing that fluvoxamine at a dose of mg/day caused a 300% increase in tolbutamide levels.18 |
Table 1 summarizes the effects of the six SSRIs on the five major CYP enzymes. As is apparent in this table, fluoxetine, fluvoxamine, and paroxetine all inhibit one or more than one CYP enzyme to a clinically substantial degree under usual dosing conditions. In contrast, citalopram, escitalopram, and sertraline do not. Moreover, fluoxetine, fluvoxamine, and paroxetine do not inhibit the same CYP enzymes. Fluoxetine and paroxetine produce substantial inhibition of CYP 2D6, whereas the other four SSRIs do not. In contrast, fluvoxamine produces substantial inhibition of CYP 1A2 and 2C19, whereas the other five SSRIs do not.19 Both fluvoxamine and fluoxetine produce substantial inhibition of 2C9/10, whereas the other SSRIs do not. From the perspective of CYP 2D6 inhibitors, fluoxetine and paroxetine belong in the same class as the antifungal, terbinafine, and the antiarrhythmic, quinidine.19
Classifying drugs in these various ways is important because such classifications have predictive powers. Obviously, therapeutic class is predictive of efficacy in a therapeutic area, even though it may not be particularly informative about the pharmacological actions of the drug. Pharmacodynamic class provides more specific and clinically useful information about pharmacological actions, even though not all drugs in a given pharmacodynamic class have been established to be useful for the same therapeutic indications. For example, sufficient evidence has never been developed for fluvoxamine to be approved by the U.S. Food and Drug Administration for labeling as an antidepressant. Sibutramine (Meridia) is a serotonin reuptake inhibitor that was never tested as an antidepressant. There are also some investigational SSRIs in development for use in the treatment of premature ejaculation that may never be tested as antidepressants.
Classification as a substantial inhibitor of a specific CYP enzyme is important when one is prescribing the inhibitor in combination with other drugs that are dependent on that enzyme for their clearance. This logic is summarized in Figure 1.
The following two equations are important in understanding the material that will be presented in this column.
Effect = affinity for site of action
x concentration at site of action
x biological variance among patients
(genetics, age, disease, environment)
(Equation 1)
Drug concentration = dosing rate / clearance
(Equation 2)
| Figure 1. How knowledge of P450 enzymes will simplify understanding of pharmacokinetic interactions | Drug A affects -> P450 enzyme X
P450 enzyme X metabolizes -> B, C, D, E, F
Therefore, Drug A affects -> B, C, D, E, F |
There are reciprocal relationships among all three variables in Equation 1. Drug concentration determines which of the potential sites of action of the drug will be engaged and to what degree. This concept was discussed at some length in a previous column.20 A figure illustrating the multiple concentration-response curves with amitriptyline was provided in another earlier column.21 Conversely, the affinity of the drug for a site of action determines what concentration of the drug must be achieved to affect that site of action to a physiologically relevant degree. The third variable in equation 1 refers to the biological differences among patients that can shift their position on the usual dose-response curve making them either more or less responsive to the drug (i.e., "sensitive" or "resistant"). Those differences may be mediated through the site(s) of action and/or through the mechanisms of pharmacokinetics.
Clinically important biological differences among patients may be genetically determined (i.e., mutations in the gene[s] coding for the site[s] of action or in the gene[s] coding for the mechanisms mediating the absorption, distribution, metabolism or elimination characteristics of the drug). They may also be due to disease- or age-related changes in the site of action or the pharmacokinetics of the drug. Biological differences among patients may also be due to environmentally determined changes resulting from the ingestion of substances, which by virtue of their presence in the body can alter the response to another drug. These substances may be dietary factors such as foods (e.g., grapefruit juice), herbs (e.g., St. Johns Wort), or drugs (e.g., antidepressants).19 This last concept is essential to an understanding of drug-drug interactions. The presence of a drug in the body is in essence a state-dependent change in the biology of the patient, which can alter the response of that patient to another drug. In fact, treatment with a specific drug can produce a phenocopy of a genetically determined difference, as will be illustrated in this column.
| Table 2. Formal in vivo studies of the effects of different SSRIs on CYP 2D6 model substrates | | SSRI | Author | N | SSRI Treatment:
dose (mg/day) x
duration (days) | Substrate | Substrate
dosing | (AUC2 – AUC1) /
AUC1
(% increase) | EMs
to
PMs | | Citalopram | Gram et al. 199323 | 8 | 40 x 10 | Desipramine | Single dose | 47% | | | Citalopram package
insert (PI)24 | NA | 40 x 22 | Metoprolol | NA | 100% | | | Escitalopram | Escitalopram PI25 | NA | 20 x 21 | Metoprolol | NA | 82% | | | Escitalopram PI25 | NA | 20 x 21 | Desipramine | NA | 100% | | | Fluoxetine | Alfaro et al. 199926 | 8 | 60 x 8* | Dextromethorphan | Single dose | | 63% | | Amchin et al. 200127 | 12 | 20 x 28* | Dextromethorphan | Single dose | | 25% | | | Bergstrom et al. 199228 | 6 | 60 x 8* | Desipramine | Single dose | 640% | | | | | 6 | 60 x 8* | Imipramine | Single dose | Imipramine 235% | | | | Preskorn et al. 199429 | 9 | 20 x 21 | Desipramine | 21 days | 380% | | | | Otton et al. 199330 | 19 | 37 ± 17 x 21 | Dextromethorphan | Single dose | | 95% | | | Maes et al. 199731 | 11 | 20 x 28 | mCPP | 7 days | 820%† | | | | Alfaro et al. 200032 | 12 | 60 x 8 | Dextromethorphan | single dose | | 42% | | Fluvoxamine | Alfaro et al. 199926 | 6 | 100 x 8 | Dextromethorphan | Single dose | | 0% | | | Spina et al. 199333 | 6 | 100 x 10 | Desipramine | Single dose | 14% | | | Paroxetine | Alderman et al. 199734 | 17 | 20 x 9 | Desipramine | 9 days | 421% | | | | Brosen et al. 199335 | 9 | 20 x 8 | Desipramine | Single dose | 364% | 78% | | | Albers et al. 199636 | 10 | 30 x 4 | Imipramine | Single dose | Imipramine 74% | | | | | | | | | Desipramine 327% | | | | Alfaro et al. 199926 | 8 | 20 x 8 | Dextromethorphan | Single dose | | 50% | | | Ozdemir et al. 199737 | 8 | 20 x 10 | Perphenazine | Single dose | 595% | | | | Alfaro et al. 200032 | 12 | 20 x 8 | Dextromethorphan | Single dose | | 83% | | | Hemeryck et al. 200038 | 8 | 20 x 6 | Metoprolol
(racemic) | Single dose | S-metoprolol 700% | | | | | | | | R-metoprolol 410% | | | Sertraline | Alderman et al. 199734 | 17 | 50 x 9 | Desipramine | 8 days | 37% | | | | Jann et al. 199539 | 4 | 50 x 7 | Desipramine | 7 days | 0% | | | | Preskorn et al. 199429 | 9 | 50 x 21 | Desipramine | 21 days | 23% | | | | Solai et al. 199740 | 13 | 50 x >5 | Nortriptyline | Chronic dosing | 14% | | | | Ozdemir et al. 199841 | 21 | 94 ±26 x 24 ±17 | Dextromethorphan | Single dose | 0% | 0% | | | Sproule et al. 199742 | 6 | 108 ±49 x 21 | Dextromethorphan | Single dose | 6% | 0% | | | Alfaro et al. 199926 | 7 | 100 x 8 | Dextromethorphan | Single dose | | 0% | | | Kurtz et al. 199743 | 6 | 150 x 8 | Imipramine | Single dose | 68% | | | | | 6 | 150 x 8 | Desipramine | Single dose | 54% | | | | Zussman et al. 199544 | 13 | 150 x 29 | Desipramine | Single dose | 70% | | | | Alfaro et al. 200032 | 12 | 100 x 8 | Dextromethorphan | Single dose | | 0% | | Abbreviations: AUC = area under the plasma concentration–time curve; EM = extensive metabolizer; PM = poor metabolizer; PI = package insert from Physician’s Desk Reference; NA = not available; mCPP = metachlorophenylpiperazine (a metabolite of nefazodone and trazodone)
*60 mg/day for 8 days is a loading dose strategy used to approximate the plasma levels of fluoxetine and norfluoxetine achieved under steady-state conditions on a dose of 20 mg/day.
†820% is based on all the data. If the two highest increases are excluded, the average was 270%.
Table adapted from Preskorn 1998.45 Copyright Preskorn 2003. |
Summary of last column
Table 2, which was first presented in Part I of this twopart series,22 presents the results of 23 prospective in vivo studies done in humans to test the relative effects of the six marketed SSRIs on the drug-metabolizing enzyme, CYP 2D6.
Two basic approaches have been used to measure the differential effects of the six SSRIs on this enzyme. The first approach involves giving the participant a drug, which is predominantly and ideally exclusively metabolized by this enzyme (i.e., a CYP 2D6 model substrate/drug), first alone, and then in the presence of an SSRI under clinically relevant dosing conditions, and measuring the increase in the levels of the model substrate/ drug when given in combination with the SSRI. The increase is typically measured as the area under the plasma-time curve (AUC). AUC1 is the condition following the administration of a given dose of the CYP 2D6 model substrate/drug in the absence of the inhibitor. AUC2 is the measure following the same dose of the CYP 2D6 model substrate/drug in the presence of the inhibitor. The formula (AUC2 - AUC1)/AUC1 represents the degree of inhibition of CYP 2D6. For the purpose of this column, this approach will be termed the AUC method.
As is clear from the data summarized in Table 2, combined treatment with either fluoxetine or paroxetine at a dose of 20 mg/day causes an average increase of 500% or more in the levels of the coadministered CYP 2D6 model substrate/drug. In contrast, the other SSRIs caused substantially lower increases: 40 mg/day of citalopram and 20 mg/day of escitalopram caused increases of 50%-100%; 100 mg/day of fluvoxamine caused a 14% increase; sertraline at doses of 50-100 mg/day caused an average increase of 15% and at doses of 150 mg/day caused an average increase of 64% (Table 2).
CYP 2D6 "poor metabolism"
In the late 1960s and early 1970s, Alexanderson et al. first discovered that some individuals were genetically deficient in the enzyme needed to metabolize tricyclic antidepressants.46 Approximately 20 years later, Gonzalez et al. isolated and identified the gene responsible for coding the human drug metabolizing enzyme, CYP 2D6.47,48 Individuals who were genetically deficient in CYP 2D6 achieved levels of CYP 2D6 preferential substrate- drugs (e.g., tricyclic antidepressants) that were four or more times higher than most people.46,49 Initially, people were divided into two categories based on their ability to metabolize and hence clear CYP 2D6 substrate- drugs. The vast majority of people were termed "extensive metabolizers" whereas those who developed unusually high levels of CYP 2D6 substrate-drugs were called "poor metabolizers." As with most genetically determined conditions, the percentage of CYP 2D6 "poor metabolizers" varied depending on the population being studied. Caucasians, particularly those of northern European heritage, had the highest percentage of CYP 2D6 "poor metabolism" (i.e., about 7%).50
Before genetic tests existed to classify people as CYP 2D6 extensive or poor metabolizers, pharmacological tests were developed using CYP 2D6 model substratedrugs such as dextromethorphan (DM).51 This cough suppressant is principally metabolized by CYP 2D6 into dextrorphan (DO). The ratio of DM to DO (i.e., DM/DO) following a standardized dose is thus a measure of the functional activity of CYP 2D6. Since individuals deficient in CYP 2D6 (i.e., poor metabolizers) cannot convert DM to DO, they will have a large DM/DO ratio; those with functional CYP 2D6 (i.e., extensive metabolizers) will have a low DM/DO ratio.
| Figure 2. Frequency distribution histograms of log dextromethorphan O-demethylation ratio (ODMR) in healthy controls before (top) and after (bottom) treatment with 40 mg/day of fluoxetine, a substantial CYP 2D6 inhibitor | 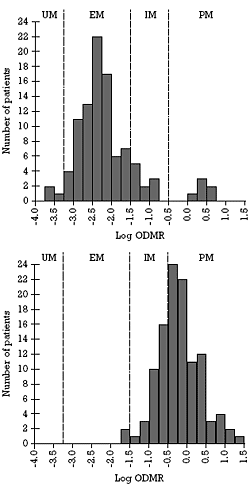 | ODMR = O-demethylation ratio of dextromethorphan;
UM = ultra-rapid metabolizer; EM = extensive metabolizer;
IM = intermediate metabolizer; PM = poor metabolizer
These histograms are modeled after data reported by Otton et al. 199330 |
This phenomenon is illustrated in the top histogram of Figure 2. In this example, there is an obvious bimodal distribution representing the EM and PM groups and a natural break at -0.5 between these two groups. This figure reflects the results typically seen with this test in a Caucasian population of northern European extraction. The size of the PM group would be smaller in other groups such as Asians, in which 1% or fewer are genetically deficient in functional CYP 2D6.50
Today, over 70 allelic variants in the CYP 2D6 gene have been identified, and genetic tests exist for these various mutations.52 Fifteen of these mutations produce a truncated form of the enzyme that is devoid of any catalytic activity. These are the mutations that account for genetically determined PM status.
Although a bit beyond the scope of this paper, the elucidation of the genetics of the CYP 2D6 gene has permitted further biologically meaningful subdivision of the data presented in Figure 2. The portion of the population in the middle of the EM distribution has two fully functional copies of the gene, including those with silent mutations (i.e., those that do not cause any known meaningful change in the structure or function of the gene product). The distribution from -1.5 to -0.5 in this figure is composed of individuals who are termed intermediate metabolizers (IMs) because they are between EMs and PMs. IMs are either heterozygous, meaning that they have one functional and one nonfunctional gene, or they are homozygous for genetic mutations that convey diminished but not absent CYP 2D6 function. Finally, there is a small population to the left side of the main population. This population is composed of individuals who are considerably more efficient metabolizers of CYP 2D6 substrates than most people and are hence termed CYP 2D6 ultra-rapid metabolizers (UMs). These individuals have a duplication of the gene and thus have twice as much activity as EMs.53 While only 1% of Caucasians are UMs, as many as 20% of Saudis and 30% of Ethiopians are UMs.54 The top histogram in Figure 2 illustrates how genetically determined biological variance among patients can shift their doseresponse curves (see Equations 1 and 2 above).
Phenocopies of CYP 2D6 PM status
The DM challenge test, as well as variations on this test using other CYP 2D6 substrates, can also be used to quantify the effect of a co-administered drug on CYP 2D6 function. This approach can complement the AUC approach. The latter gives the more quantitatively relevant result, while the DM approach gives a biologically determined dichotomy (i.e., EM versus PM status). The AUC approach provides a measure of the mean change in the levels of the CYP 2D6 substrate and the standard deviation provides a measure of the variance in the effect. The DM approach provides a measure of what percentage of the EM population becomes a phenocopy of genetic deficiency of the enzyme.
The bottom histogram of Figure 2 illustrates this last point by showing what happens to the EM population when they are treated with a substantial CYP 2D6 inhibitor. The entire population is shifted to the right, meaning that CYP 2D6 activity and clearance slowed in everyone in the population, including the IMs, EMs, and UMs. In approximately 90% of the population, the degree of CYP 2D6 inhibition was sufficient for them to cross the biologically relevant threshold on this test to qualify as PMs (i.e., having no functional activity of this enzyme).
The last column of Table 2 presents the results of such studies with four of the six SSRIs. The results of this type of study have not been reported for citalopram and escitalopram but would be expected to be closer to the results for fluvoxamine and sertraline than for fluoxetine and paroxetine, based on the results from the AUC type of study. Fluoxetine, at a dose of 20 mg/day, converted 25% to 63% of EMs to PM status in three separate studies, for an average of 43%. In one study, fluoxetine at 40 mg/day converted 95% from EMs to PMs. Paroxetine, at a dose of 20 mg/day, converted 50% to 83% of EMs to PM status in three separate studies, for an average of 70%. Fluvoxamine, at a dose of 100 mg/day in one study, converted no EMs to PM status. Sertraline, at a dose of 100 mg/day in four separate studies, converted no EMs to PM status. These results are fully consistent with the results obtained using the AUC approach.
Factors determining the magnitude of the effect
There are several factors that determine the magnitude of the effect seen in the studies summarized in Table 2. These factors are those outlined in Equations 1 and 2. In these studies, the effect in equation 1 is the change in either the concentration of the CYP 2D6 model substrate/drug (i.e., the AUC studies) or the percentage of the EM population converted to PM status (i.e., the DM challenge studies). In other words, the effect is the degree of reduction in the clearance of the CYP 2D6 model substrate/drug (equation 2) because:
Clearance = dosing rate / drug concentration
(Equation 2 rearranged)
Equation 1 then contains all of the elements needed to understand the differential effects of these four SSRIs. The first and most important variable is the difference among these drugs in their unintended and undesired ability to inhibit CYP 2D6 (variable 1 in equation 1). That ability can be measured in vitro as the kinetic rate constant (i.e., Ki) for the drugs ability to inhibit this enzyme. The second variable is the dose of the inhibitor, because that determines the concentration of the inhibitor at the enzyme (variable 1 in equation 2). That is germane to the finding that 95% of EMs were converted to PM status by fluoxetine at a dose of 40 mg/day versus an average of 43% at a dose of 20 mg/day. As the dose of the inhibitor goes up, the concentration of the inhibitor goes up (equation 2) and the degree of enzyme inhibition goes up, meaning that a larger percentage of the population crosses the threshold to qualify for PM status (i.e., the DM phenotyping studies) and there is a greater increase in the concentration of the CYP 2D6 model substrate/drug (i.e., the AUC studies).
Obviously, the magnitude of the effect is also related to the persons level of enzyme activity before the inhibitor was given and the persons ability to clear the inhibitor (variable 3 in equation 1). To further elaborate on this last point, everyone did not cross the threshold to PM status in all of the studies with either fluoxetine or paroxetine or in the bottom histogram in Figure 2, even though everyone was shifted to the right (i.e., reduced CYP 2D6 activity). Those with higher intrinsic activity of the enzyme would need higher concentrations (higher doses) of the inhibitor to achieve PM status (i.e., an example of variable 3 modifying variable 1). Conversely, those with greater intrinsic ability to clear the inhibitor would need higher doses of the inhibitor to achieve a concentration of the inhibitor capable of fully inhibiting the enzyme (i.e., an example of variable 3 modifying variable 2 in equation 1). Those with both higher intrinsic activity of the enzyme and higher ability to clear the inhibitor would have two biological protections against achieving PM status as a result of treatment with either 20 mg/day of fluoxetine or paroxetine.
Given this explanation, it is obvious that a lower AUC effect and a smaller percentage of EM to PM conversions would be obtained if the study were conducted only in UM individuals or only in individuals with the ability to rapidly clear fluoxetine or paroxetine.55 For the same reasons, no effect would be observed if the study were conducted in genetically determined PMs because they do not have any enzyme to be inhibited so their clearance of CYP 2D6 substrates would not change because they have no ability to clear substrates via this pathway.
Parenthetically, the clearance of fluoxetine and paroxetine are not dependent on CYP 2D6 under usual dosing conditions because these drugs saturate this enzyme. At low concentrations, paroxetine is preferentially cleared via CYP 2D6 but its clearance becomes dependent on another CYP enzyme, most likely CYP 3A, as its concentration increases.16,17 That is why its half-life changes from 10 hours following a single dose of 20 mg to approximately 20 hours at steady-state on 20 mg/day.56 This is an illustration of nonlinear pharmacokinetics and is due to the inhibition of CYP 2D6 as paroxetine accumulates (i.e., autoinhibition of metabolism which is the flip side of autoinduction seen with drugs such as carbamazepine).
The bigger picture
While these results are important in their own right, they also illustrate a more fundamental biological paradigm. CYP 2D6 is but one of thousands of biologically relevant regulatory proteins (i.e., gene products). The explanation for the variance in the effect mediated by a drugs action on any other regulatory protein would follow the same lines as the explanation presented above. In each case, the effect would be dependent on the site of action (i.e., the regulatory protein), the concentration of the drug affecting that site of action (i.e., the perpetrator drug), whether that drug is an agonist, antagonist, or inverse agonist, and the biological variance among different patients that can shift their concentration (dose)- response curve to make them more or less sensitive to the effects of the perpetrator drug. That biological variance can be due to any of the factors mentioned in equation 1: genetics, age, disease, and internal milieu (e.g., dietary substances, concomitant drug therapy).
All genes can have variants that change the confirmation and hence the intrinsic activity of the regulatory protein and/or the ability of drugs to bind and affect the protein. For example, a distribution could exist in the intrinsic activity of the dopamine-2 (D-2) receptor analogous to the distribution seen in the top histogram in Figure 2. There could be individuals with a mutation in the D-2 gene analogous to the duplication of the CYP 2D6 gene seen in UMs. Such a mutation could convey increased intrinsic D-2 activity or possibly reduced binding affinity for D-2 blockers (e.g., haloperidol). These individuals would be resistant to the effects of D- 2 blockers as would individuals who are able to rapidly clear a specific D-2 blocker (whether genetically determined or acquired as a result of concomitant drug therapy with a specific enzyme inducer).
Conversely, individuals with a mutation in the D-2 gene that conveys lower than usual intrinsic activity or greater binding affinity for D-2 blockers would be analogous to CYP 2D6 IMs. These individuals would be more sensitive to the effects of D-2 blockers (i.e., shifted to the left). The same would be true for individuals who have a reduced ability to clear a specific D-2 blocker (whether genetically determined or acquired as a result of concomitant drug therapy with a specific enzyme inhibitor). If the number of D-2 receptors decreased with age, then age could become a variable that shifts the individual over time in terms of their sensitivity to D-2 blockers, although their absolute sensitivity might still be dependent on their original status. Finally, a disease state such as Parkinsons disease, in which there is a loss of dopamine cells in the substantia nigra, could shift the patients ability to compensate for D-2 blockade. That is why patients with Parkinsons disease are more sensitive to the extrapyramidal effects of D-2 antagonists such as haloperidol and even the D-2 blockade produced by newer "atypical" antipsychotics.
These points about clearance modifying the effects of a drug form the conceptual underpinning for therapeutic drug monitoring (TDM). TDM allows the clinician to test for one reason why a specific patient may be either sensitive or resistant to the usual effects of a given dose of a given psychiatric medication. Given the large number of unknown variables that could account for variance in the response to most psychiatric medications, TDM can provide definitive, quantitative data on one of these variables (i.e., the ability of the patient to clear the effector drug). It is thus surprising that TDM is not used more often in clinical practice as a cost-effective and scientific approach to dose adjustment in a specific patient.
Summary
While CYP 2D6 inhibition is specifically important to patients who are being treated concomitantly with CYP 2D6 inhibitors and CYP 2D6 substrates, the bigger picture is even more important conceptually. This two-part series of columns ties together the topics of the different sections of columns on my website (www.preskorn. com). Section 3 outlines the fundamental understanding of dose (concentration)-response curves, which is dependent on the factors discussed above. Section 4 reviews the emerging understanding of genetic variables that can shift dose (concentration)-response curves and discusses the ability to identify previously unknown regulatory proteins (variable 1 in equation 1), which in turn can be targets for new drug discovery. Section 1 provides real-life case examples illustrating how various types of biological variance (whether inherited or acquired) can affect treatment outcome to a clinically meaningful degree. Section 2 illustrates how a therapeutic class (e.g., antidepressants) composed of pharmacologically diverse drugs can be taken and reclassified according to the site or mechanism of action (i.e., the first variable in equation 1) into pharmacologically defined subgroups in order to better understand and predict the clinical effects.
References
- Preskorn SH, Do you believe in magic? J Pract Psychiatry Behav Health 1997;3:99-103
- Preskorn SH, I don’t see ’em. J Prac Psych Behav Hlth. 1997; 3:302-307
- Preskorn SH, A message from Titanic. J Prac Psych Behav Hlth. 1998;4:236-242
- Preskorn SH, A tale of two patients. J Pract Psychiatry Behav Health 1999;5:160-164
- Preskorn SH, Silkey B, Multiple medications, multiple considerations. Journal of Psychiatric Practice 2001;7:48-52
- Preskorn SH, The human genome project and drug discovery in psychiatry: Identifying novel targets. Journal of Psychiatric Practice 2001;7:133-7
- Preskorn SH, Drug approvals and withdrawals over the last 60 years. Journal of Psychiatric Practice 2002;7:410
- Preskorn SH, Fatal drug-drug interaction as a differential consideration in apparent suicides. Journal of Psychiatric Practice 2002;8:233-8
- Preskorn SH, Clinical pharmacology case conference: A suicide attempt? Journal of Psychiatric Practice 2002;8:306-10
- Preskorn SH, Polypharmacy in a patient with refractory major depression: Part I. The case. Journal of Psychiatric Practice 2002;8:370-6
- Preskorn SH, Polypharmacy in a case of refractory major depression: Part II: Implications for clinical management. Journal of Psychiatric Practice 9(1), January 2003
- Preskorn SH, The human genome project and modern drug development in psychiatry. Journal of Psychiatric Practice 2000;6:272-276
- Preskorn SH, Marooned: Only one choice. J Prac Psych and Behav Hlth 1998;4:110-4
- Preskorn SH, Outpatient management of depression: A guide for the primary-care practitioner, 2nd Edition. Caddo, OK: Professional Communications; 1999
- Preskorn SH, Clinical Pharmacology of Selective Serotonin Reuptake Inhibitors. Caddo, Okla: Professional Communications, Inc; 1996
- Harvey AT, Preskorn SH, Cytochrome P450 enzymes: interpretation of their interactions with selective serotonin reuptake inhibitors. Part I. J Clin Psychopharmacol. 1996;16:273-285
- Harvey AT, Preskorn SH, Cytochrome P450 enzymes: interpretation of their interactions with selective serotonin reuptake inhibitors. Part II. J Clin Psychopharmacol. 1996;16:345-355
- Madsen H, Enggaard TP, Hansen LL, et al., Fluvoxamine inhibits the CYP2C9 catalyzed biotransformation of tolbutamide. Clin Pharmacol Ther 2001;69:41-7
- Shad MU, Preskorn SH, Antidepressants. In: Levy RH, Thummel KE, Trager WF, et al., eds. Metabolic drug interactions. Philadelphia: Lippincott, Williams & Wilkins; 2000:563-77
- Preskorn SH, Defining "Is." J Pract Psychiatry Behav Health 1999;5:224-8
- Preskorn SH, What happened to Tommy? J Pract Psychiatry Behav Health 1998;4:363-7
- Preskorn SH, Reproducibility of the in vivo effect of the selective serotonin reuptake inhibitors on the in vivo function of cytochrome P450 2D6: An update (part I) Journal of Psychiatric Practice 9(2), March 2003
- Gram LF, Hansen MGJ, Sindrup SH, et al, Citalopram: interaction studies with levomepromazine, imipramine, and lithium. Ther Drug Monit. 1993;15:18-24
- Citalopram package insert. Physicians’ Desk Reference. Montvale, NJ: Thomson PDR; 2003: 1344-7
- Escitalopram package insert. Physicians’ Desk Reference. Montvale, NJ: Thomson PDR; 2003: 3532-5
- Alfaro CL, Lam YWF, Simpson J, et al, CYP2D6 Status of extensive metabolizers after multiple-dose fluoxetine, fluvoxamine, paroxetine or sertraline. J Clin Pharmacol 1999;19: 155-63
- Amchin J, Ereshefsky L, Zarycranski W, et al, Effect of venlafaxine versus fluoxetine on metabolism of dextromethorphan, a CYP2D6 probe. J Clin Pharmacol 2001;41:443-51
- Bergstrom RF, Peyton AL, Lemberger L, Quantification and mechanism of the fluoxetine and tricyclic antidepressant interaction. Clin Pharmacol Ther. 1992;51:239-248
- Preskorn SH, Alderman J, Chung M, Harrison W, Messig M, Harris S, Pharmacokinetics of desipramine coadministered with sertraline or fluoxetine. J Clin Psychopharmacol. 1994;14:90-98
- Otton SV, Wu D, Joffe RT, Cheung SW, Sellers EM, Inhibition by fluoxetine of cytochrome P-450 2D6 activity. Clin Pharm Therap. 1993;53:401-409
- Maes M, Westenberg H, Vandoolaeghe E, et al, Effects of trazodone and fluoxetine in the treatment of major depression: Therapeutic pharmacokinetic and pharmacodynamic interactions through formation of meta-chlorophenylpiperazine. J Clin Psychopharmacol 1997;17:358-64
- Alfaro CL, Lam YWF, Simpson J, et al, CYP2D6 Inhibition by fluoxetine, paroxetine, sertraline, and venlafaxine in a crossover study: Intraindividual variability and plasma concentration correlations. J Clin Pharmacol 2000;40:58-66
- Spina E, Pollicino AM, Avenoso A, Campo GM, Perucca E, Caputi AP, Effect of fluvoxamine on the pharmacokinetics of imipramine and desipramine in healthy subjects. Ther Drug Monit. 1993;15:243-246
- Alderman J, Preskorn S, Greenblatt D, et al, Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 1997;17:284-91
- Brøsen K, Hansen JG, Nielsen KK, Sindrup SH, Gram LF, Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur J Clin Pharmacol. 1993;44:349-355
- Albers LJ, Reist C, Helmeste D, Vu R, Jang SW, Paroxetine shifts imipramine metabolism. Psychiatry Research. 1996; 59:189-196
- Ozdemir V, Naranjo CA, Herrmann N, et al, Paroxetine potentiates the central nervous system side effects of perphenazine: Contribution of cytochrome P4502D6 inhibition in vivo. Clin Pharmacol Ther 1997;62:334-47
- Hemeryck A, Lefebvre RA, De Vriendt C, et al, Paroxetine affects metoprolol pharmacokinetics and pharmacodynamics in healthy volunteers. Clin Pharmacol Ther 2000;67:283-91
- Jann MW, Carson SW, Grimsley SR, Erikson SM, Kumar A, Carter JG, Lack of effect of sertraline on the pharmacokinetics and pharmacodynamics of imipramine and its metabolites. Clin Pharm Therap. 1995;57:207. Abstract
- Solai LK, Mulsant BH, Pollock BG, et al, Effect of sertraline on plasma nortriptyline levels in depressed elderly. J Clin Psychiatry 1997;58:440-3
- Ozdemir V, Naranjo CA, Hermann N, et al, The extent and determinants of changes in CYP2D6 and CYP1A2 activities with therapeutic doses of sertraline. J Clin Psychopharmacol 1998;18:55-61
- Sproule B, Otton SV, Cheung SW, et al, CYP2D6 inhibition in patients treated with sertraline. J Clin Psychopharmacol 1997; 17:102-6
- Kurtz D, Bergstrom RF, Goldberg MJ, et al, The effect of sertraline on the pharmacokinetics of desipramine and imipramine. Clin Pharmacol Ther 1997;62:145-56
- Zussman BD, Davie CC, Fowles SE, et al, Sertraline, like other SSRIs, is a significant inhibitor of desipramine metabolism in vivo. Br J Clin Pharmacol. 1995;39:550-551
- Preskorn SH, Debate resolved: there are differential effects of serotonin selective reuptake inhibitors on cytochrome P450 enzymes. J Psychopharmacol. 1998;12:S89-S97
- Alexanderson B, Sjoqvist F, Individual differences in the pharmacokinetics of monomethylated tricyclic antidepressants: Role of genetic and environmental factors and clinical importance. Ann NY Acad Sci 1971;179:739-51
- Gonzalez FJ, Skoda RC, Kimura S, et al, Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442-446
- Gonzalez FJ, Vilbois F, Hardwick JP, et al, Human debrisoquine 4-hydroxylase (P450IID1): cDNA and deduced amino acid sequence and assignment of the CYP2D locus to chromosome 22 Genomics 1988;2:174-9
- Asberg M, Individualization of treatment with tricyclic compounds. Med Clin North Am 1974;58:1083-91
- Weber WW., Populations and genetic polymorphisms. Mol Diagn 1999;4:299-307
- Kohler D, Hartter S, Fuchs K, et al, CYP2D6 genotype and phenotyping by determination of dextromethorphan and metabolites in serum of healthy controls and of patients under psychotropic medication. Pharmacogenetics 1997;7:453-61
- Bertilsson L, Dahl ML, Dalen P, et al., Molecular genetics of CYP2D6: Clinical relevance with focus on psychotropic drugs. Br J Clin Pharmacol 2002;53:111-22
- Lovlie R, Daly AK, Molven A, et al., Ultrarapid metabolizers of debrisoquine: Characterization and PCR-based detection of alleles with duplication of the CYP2D6 gene. FEBS Lett 1996;392:30-4
- McLellan RA, Oscarson M, Seidegard J, et al, Frequent occurrence of CYP2D6 gene duplication in Saudi Arabians. Pharmacogenetics 1997;7:187-91
- Laine K, Tybring G, Hartter S, et al., Inhibition of cytochrome P4502D6 activity with paroxetine normalizes the ultrarapid metabolizer phenotype as measured by nortriptyline pharmacokinetics and the debrisoquin test. Clin Pharmacol Ther 2001;70:327-35
- Kaye CM, Haddock RE, Langley PF, et al, A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatr Scand. 1989;80(supp 350):60-75
|



